Synthesis and characterization of the Ce 1 -Ru n /NC. The united catalyst of oxophilic Ce single atoms and fully-exposed small Ru nanoclusters was prepared on a N functionalized XC-72 carbon support (indicated as Ce1-Run/NC, see Figure S1-2 for details). As a control, pure Ru nanoclusters and Ce single atoms were also prepared on the NC support (denoted as Run/NC and Ce1/NC, respectively) using a similar method. The loading amount of Ce in Ce1/NC (Fig. 1a) was measured to be 0.03wt.% by an inductively coupled plasma optical emission spectrometer (ICP-OES). Due to the low Ce loadings, only the broad X-ray diffraction (XRD) peaks of carbon support23 were observed for the Ce1/NC as displayed in Figure S3. In addition, the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of the Ce1/NC in Figure S4-6 excluded any particles in it. It was further unveiled by the aberration-corrected HAADF-STEM (AC HAADF-STEM) measurement that Ce species were atomically dispersed in Ce1/NC (Fig. 1b-d) and corresponding energy-dispersive X-ray spectrometry (EDS) elementary mapping images in Fig. 1e-g also confirmed the uniform Ce dispersion.
Likewise, we have characterized the Run/NC (Fig. 1h) and the Ce1-Run/NC (Fig. 1o) via the microscopic techniques. As presented by the AC HAADF-STEM images of the Run/NC in Fig. 1i-j, fully-exposed Ru nanoclusters with an average particle size of 1.1 ± 0.3 nm were evenly dispersed on the NC support. The EDS elementary mapping images of a small nanocluster in Run/NC (Fig. 1k-n) revealed its Ru element nature. The AC HAADF-STEM images of the Ce1-Run/NC in Fig. 1p-q showed that the mean particle size of the fully-exposed Ru nanoclusters in it was 1.0 ± 0.2 nm. Meanwhile, Ce species were atomically dispersed around the fully-exposed Ru nanoclusters. Corresponding EDS elementary mapping images in Fig. 1r-u also indicated uniform dispersion of the fully-exposed Ru nanoclusters and the Ce single atoms in Ce1-Run/NC. No diffraction peaks of Ru were identified for the Run/NC and the Ce1-Run/NC as shown in Figure S7 because of their ultrasmall Ru nanoclusters. The Ru loading amounts of the Ce1-Run/NC and the Run/NC were both 1wt.% while the Ce weight loading in Ce1-Run/NC was 0.03wt.% as determined by the ICP-OES measurements.
The coordination environment of Ru and Ce in Ce1/NC, Run/NC and Ce1-Run/NC was further examined by the X-ray absorption fine-structure (XAFS) measurement. Figure 2a showed the near-edge XAFS spectra (XANES) at Ru K-edge of the Ce1-Run/NC, Run/NC and reference Ru foil and RuO2. It was displayed by the enlarged Ru K-edge XANES spectra in the inset of Fig. 2a that the edge absorption energies of Ce1-Run/NC and Run/NC were between Ru foil and RuO2, which demonstrated the oxidation state of the fully-exposed Ru nanoclusters in Ce1-Run/NC and Run/NC was between 0 and + 4. The formation of positively charged Ru nanoclusters in Ce1-Run/NC and Run/NC was due to the strong N-Ru coordination through which Ru electrons could be easily transferred to connecting N of much larger electronegativity than Ru24, hence leading to the increase of oxidation state of Ru nanoclusters of them. The edge energy of the Ce1-Run/NC showed an obvious negative shift relative to the Run/NC and thus an enhancement of its Ru electron density,25–26 which possibly derived from Ce electron donation. To check this assumption, the Bader charge analysis were further conducted. It was demonstrated in Figure S8 that electrons could be facilely transferred from Ce to Ru with a net electron transfer number of 0.05, which agreed well with the XANES data.
The Fourier transforms of the Ru K-edge extended XAFS (EXAFS) oscillations in the R space of the Ce1-Run/NC and the Run/NC in Fig. 2b presented a main coordination peak below 2 Å, matching to the Ru-N coordination.27 The Ru-Ru coordination peak at about 2.7 Å was also identified for both of the Ce1-Run/NC and the Run/NC. However, the peak intensity of Ru-Ru coordination was largely reduced relative to Ru-N coordination, implying their significant difference in coordination numbers.28 The EXAFS data fitting of the Ce1-Run/NC and the Run/NC in Fig. 2c-d and Figure S9-10 then provided the exact Ru-Ru and Ru-N coordination information of them. As summarized in Table S1, the Ru-N coordination ratio was largely increased compared with the Ru-Ru coordination both for the Ce1-Run/NC and the Run/NC. The enhanced Ru-N coordination ratio was a feature for N-stabilized small Ru nanoclusters because the particle downsizing of Ru significantly decoupled the Ru-Ru bond and strengthened the Ru-N coordination.26 This result was also supported by the EXAFS wavelet transform (WT) results. As displayed in Fig. 2e, the Ru-N coordination intensity was obviously stronger than the Ru-Ru coordination both for the Ce1-Run/NC and the Run/NC. The Ce LIII-edge XANES and EXAFS spectra of the Ce1-Run/NC in Fig. 2f-g and Figure S11-13 again confirmed the single atom nature of Ce in Ce1-Run/NC, which was consistent with the above AC HAADF-STEM observation. In addition, the electron transfer between Ce single atoms and Ru nanoclusters in Ce1-Run/NC was further evidenced by the Ru 3p XPS spectra in Fig. 2h, where the Ru0 binding energy peak of the Ce1-Run/NC was slightly negatively shifted compared with the Run/NC because of the electron back-donation from Ce to Ru.
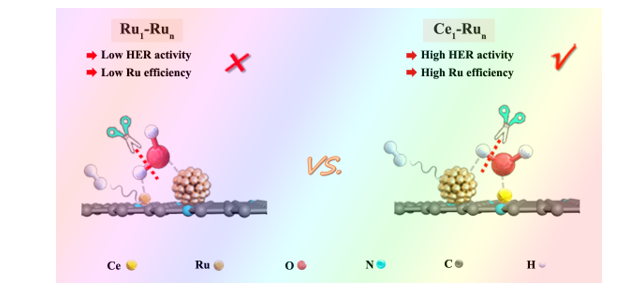
Electrocatalytic evaluations for alkaline HER. According to reported findings,29 water dissociation on the surface of catalyst was largely dependent on corresponding OH adsorption strength. Strong OH binding was beneficial to water molecules’ polarization and elongation and commonly promoted water dissociation. Thus, we have firstly conducted the cyclic voltammetry (CV) measurements in 1.0 M KOH solution to explore the OH adsorption strength on the Run/NC and the Ce1-Run/NC catalysts, respectively, before electrochemical alkaline HER evaluations. Figure 3a displayed that the OH-adsorption peak intensity around 0.60 to 1.00 V30 of the Ce1-Run/NC was much stronger than the Run/NC, indicating intensified OH adsorption on the Ce1-Run/NC over the Run/NC thanks to Ce single atoms promoting. As a control, we have also prepared a CeO2 nanoparticle integrated Run catalyst on NC support (Run-CeO2/NC, Figure S14-18) with similar Ru nanoclusters to Run/NC and Ce1-Run/NC while the Ce sites therein were bonded with O. It was found that the OH-adsorption peak intensity of the Run-CeO2/NC was markedly decreased relative to the Ce1-Run/NC due to the O blocking of Ce sites. These results indicated that OH was prone to bond with Ce single atoms of the Ce1-Run/NC catalyst as vividly depicted in Fig. 3b.
To further confirm this, we have performed the in situ Raman measurements in 1.0 M KOH electrolyte under real alkaline HER conditions (Figure S19). As presented in Fig. 3c, the Raman vibration peaks at 1533 cm-1 and 1390 cm-1 of the Ce1-Run/NC catalyst were gradually intensified with the increase of reaction potentials during alkaline HER. By contrast, the Run/NC catalyst merely showed the vibration peaks for the D band and G band of carbon support (Fig. 3d) under the same testing conditions.31 Since there was no record of the two Raman vibration peaks of the Ce1-Run/NC catalyst at 1533 cm-1 and 1390 cm-1, we have further conducted the theoretical simulations. Through theoretical fitting of the Raman spectrum of the Ce1-Run/NC catalyst before alkaline HER test (Fig. 3e), we found that the Raman vibration peaks at 1533 cm-1 and 1390 cm-1 were assigned to the Ce-N stretching vibration but the two peaks were very weak before alkaline HER test. Thus, it was reasonable to assume that the strengthening of the Raman vibration peaks at 1533 cm-1 and 1390 cm-1 was related to OH or H produced during the alkaline HER process. To this end, we have examined both OH and H effects on the Ce-N stretching vibration. Strikingly, upon introducing OH to the Ce-N sites of the Ce1-Run/NC catalyst, corresponding Raman peak intensities at 1533 cm-1 and 1390 cm-1 were significantly strengthened as shown in Fig. 3f, agreeing well with the in situ Raman spectra in Fig. 3b. By contrast, it was manifested in Figure S20 that H had a negligible impact on intensifying the Ce-N stretching vibration at these positions. Evidently, OH was more susceptible to bond with the Ce single atoms in Ce1-Run/NC during alkaline HER and in turn made the fully-exposed Ru nanoclusters therein serve as hydrogen evolution centers, hence realizing the active site reverse relative to the Ru1-Run catalyst.
The Ce1-Run/NC and other control catalysts were then employed for electrochemical alkaline HER evaluations. As expected, the Ce1-Run/NC exhibited much enhanced catalytic activity than other catalysts as indicated by their linear sweep voltammetry (LSV) curves in Fig. 4a. To make it clear, the current density of the Ce1-Run/NC at − 0.05 V was up to 30 mA cm-2, which was 2.3 times and 4.6 times that of the 20wt.% Pt/C and the Run/NC as demonstrated in Fig. 4b. When the reaction potential reached − 0.15 V, the current density of the Ce1-Run/NC was much improved to 281 mA cm-2 and became 3.5 times that of the 20wt.% Pt/C and 8.5 times that of the Run/NC. Moreover, the mass activity of the Ce1-Run/NC was even larger than the 20wt.% Pt/C as shown in Figure S21. Figure 4c disclosed that the Tafel slope of the Ce1-Run/NC was the lowest among these catalysts, thus endowing the Ce1-Run/NC with fast reaction kinetics for hydrogen evolution. Additionally, the smallest charge transfer resistance (Rct) of the Ce1-Ru/NC as shown in Figure S22 provided further evidence for its rapid HER kinetics. To examine the intrinsic activity of these catalysts, we have conducted the turnover frequency (TOF) measurements in the potential range from − 0.01 V to − 0.06 V. It was found that the TOF values of the Ce1-Run/NC and the Run/NC were obviously higher than the 20wt.% Pt/C as shown in Fig. 4d. Moreover, with the assistance of Ce single atoms, the TOF value of the Ce1-Run/NC was significantly larger than the Run/NC. By contrast, the Run-CeO2/NC showed obviously reduced TOF value with respect to the Ce1-Run/NC, which possibly derived from its weak OH adsorption strength as demonstrated by the aforementioned CV results in Fig. 3a.
Figure 4e and Figure S23 presented a comparison of the alkaline HER mass activity between the Ce1-Run/NC and other reported Ru-based catalysts. Obviously, the Ce1-Run/NC catalyst outperformed all the Ru-based alkaline HER catalysts to date to the best of our knowledge in terms of mass activity both at − 0.05 V and − 0.1 V (corresponding data were also provided in Table S2 and S3). Stability measurement of alkaline HER was commonly conducted at the current density of 10 mA cm-2 to fulfill the requirement of electrochemical performance at a device level.32 However, the current density should be no less than 100 mA cm-2 in order to meet the commercial standard for industrial alkaline HER.33 Therefore, we have measured the catalytic stability of the Ce1-Run/NC and the commercial 20wt.% Pt/C at a high current density of 150 mA cm-2. As demonstrated in Fig. 4f, the Ce1-Run/NC catalyst displayed excellent catalytic stability for 100 hours at such high current density while the 20wt.% Pt/C showed obvious catalytic stability degrading in less than 3 hours, displaying great potential of the Ce1-Run/NC catalyst for practical applications.
Theoretical investigations for the Ce 1 -Ru n /NC catalyst’s superior alkaline HER activity. The first-principle density functional theory (DFT) calculations with spin-polarization were further carried out to explore the origin of the excellent alkaline HER activity of the Ce1-Run/NC catalyst. The structural models of the Ce1-Run/NC, Run/NC and Ce1/NC were built based on the EXAFS fitting data, wherein Ce single atom was coordinated with six N atoms and Ru nanocluster was stabilized by four N atoms on the NC support. All the structure details and model files of the Ce1-Run/NC, Run/NC and Ce1/NC catalysts were provided in supporting information (Figure S24-26). It was found that the Ce1/NC showed a huge Gibbs free energy barrier for water dissociation as displayed in Figure S27-28, which explained its low reactivity for the alkaline HER (Fig. 4a). Figure 5a showed that the Ce1-Run/NC catalyst possessed a micro-electric field due to the notable difference in the electrostatic potential distribution between Ce single atoms and fully-exposed Ru nanoclusters. Consequently, the polarized water molecules could be easily elongated upon contacting the Ce1-Run/NC catalyst. Notably, the O-H bond of water was significantly elongated from 0.98 Å to 1.04 Å when adsorbed on the Ce1-Run/NC catalyst, hence efficiently favoring water activation.
In theory, water had two possible activation patterns on the Ce1-Run/NC catalyst: i) the OH groups bond with Ce single atoms while the H atoms bond with Ru nanoclusters (Ce1OH-RunH route); and ii) the H atoms bond with Ce single atoms while the OH groups bond with Ru nanoclusters (Ce1H-RunOH route). Therefore, we have performed DFT calculations for both routes. As displayed in Fig. 5b, the Ce1H-RunOH route was endothermic by 0.6 eV while the Ce1OH-RunH route was particularly exothermic by 4.5 eV. The sharply different thermodynamics of the Ce1H-RunOH route and the Ce1OH-RunH route was most likely derived from the distinct oxophilicity of Ru and Ce. Compared with the d-block Ru, the f-block Ce was much more oxophilic34 by means of which Ce was more prone to bond with OH instead of H, hence making the Ce1OH-RunH route more thermodynamically preferred.
As far as the Ce1-Run/NC and the Run/NC were concerned, the exothermic energy of the Ce1-Run/NC (4.5 eV, Ce1OH-RunH route) was 9 times that of the Run/NC (0.5 eV) in water activation process, revealing the Ce1-Run/NC was more thermodynamically favorable to dissociate water than the Run/NC. Besides, the Gibbs free energy barriers for breaking OH-H bond of water over the Ce1-Run/NC catalyst was also lower than the Run/NC. Therefore, the Ce1-Run/NC catalyst was both thermodynamically and kinetically beneficial to promote water dissociation. After water dissociation, OH was energetically bonded with the oxophilic Ce single atoms while H was adsorbed on the fully-exposed Ru nanoclusters. To gain insight into the hydrogen formation and the OH– desorption over the Ce1-Run/NC and the Run/NC catalysts, we have further carried out calculations for H binding strength (∆GH*) and OH– desorption energy barriers of them.
Because of the electron enriching of Ru in the Ce1-Run/NC compared with the Run/NC as revealed by the aforementioned XANES and XPS results, the calculated d-band center of Ru in the Ce1-Run/NC (–1.48 eV) displayed an obvious downshift compared with the Run/NC (–1.43 eV) as shown in Fig. 5c and d. According to previous reports,29 the low reactivity of Ru metal toward HER was usually ascribed to its too strong H adsorption that hindered H transfer and H2 formation. The Ru d-band center downshift of the Ce1-Run/NC would facilely weaken H binding strength35 to enable more facile H transfer. As shown in Fig. 5e, the ∆GH* of the Ce1-Run/NC was obviously closer to the optimum than the Run/NC, which made the Ce1-Run/NC more efficient for hydrogen evolution. Furthermore, the electron localization function (ELF) analysis (inset of Fig. 5e) disclosed that a weaker degree of electron localization in the Ru-H bonding region was identified for the Ce1-Run/NC than the Run/NC, thus weakening the hydrogen binding strength thereon for more favorable hydrogen production. On the other hand, the OH– desorption from the Ce1-Run/NC was also greatly favored compared with the Run/NC in view of its lower OH– desorption energy barrier (1.0 eV) as indicated in Fig. 5b.
In conclusion, we report a united catalyst of oxophilic Ce single atoms and fully-exposed Ru nanoclusters on a N functionalized carbon support that facilely reverses the hydrogen evolution centers to the more efficient Ru nanoclusters during alkaline HER, hence attaining significantly improved HER activity and Ru atom efficiency. Notably, the mass activity of the Ce1-Run/NC surpassed all the Ru-based alkaline HER catalysts to date, coupled with excellent durability. Extensive experiment data and theoretical calculations well confirmed the active site reverse over the Ce1-Run/NC catalyst for more efficient alkaline hydrogen evolution. This unexpected finding possibly provides new insights into designing more cost-effective alkaline HER catalyst.
{kind=link}