The current severe situation of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has not been reversed and posed great threats to global health. Therefore, there is an urgent need to find out effective antiviral drugs. The 3-chymotrypsin-like protease (3CLpro) in SARS-CoV-2 serve as a promising anti-virus target due to its essential role in the regulation of virus reproduction. Here, we report an improved integrated approach to identify effective 3CLpro inhibitors from effective Chinese herbal formulas. With this approach, we identified the 5 natural products (NPs) including narcissoside, kaempferol-3-O-gentiobioside, rutin, vicenin-2 and isoschaftoside as potential anti-SARS-CoV-2 candidates. Subsequent molecular dynamics simulation additionally revealed that these molecules can be tightly bound to 3CLpro and confirmed effectiveness against COVID-19. Moreover, kaempferol-3-o-gentiobioside, vicenin-2 and isoschaftoside were first reported to have SARS-CoV-2 3CLpro inhibitory activity. In summary, this optimized integrated strategy for drug screening can be utilized in the discovery of antiviral drugs to achieve rapid acquisition of drugs with specific effects on antiviral targets.
Research article
An integrated method for optimized identification of effective natural inhibitors against SARS-CoV-2 3CLpro
https://doi.org/10.21203/rs.3.rs-314304/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this older preprint version
Herbal medicine
Natural products
Pipeline of drug screening
COVID-19
3-chymotrypsin-like protease
Coronavirus disease 2019 (COVID-19) is an infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which can lead to various symptoms including fever, cough, fatigue, shortness of breath, and loss of smell and taste [1]. To date, more than 112.0 million persons have been infected into COVID-19 across 192 countries or regions which resulted into 2,484,988 deaths [2]. Thus it will be urgent to seek for the effective treatment against COVID-19.
Since the outbreak of pneumonia, a series of compounds have been developed to treat COVID-19 [3]. At present, mainstream antivirus research has been conducted on the mechanism of virus replication, and many effective compounds have been discovered. A series of antivirals being tested for SARS-CoV-2 such as PF-07304814 [4], remdesivir [5], GC376 [6], apilimod [7], nelfinavir [8] and quinacrine [9], displayed high antiviral activity in vitro. Some of them, such as remdesivir [10], favipiravir [11] and PF-07304814 are also under clinical investigation. However, no drugs have shown outstanding therapeutic effects in clinical trials. As the epidemic situation remains grim, there is still an urgent need for effective methods to discover valid antiviral drugs.
In addition to the mentioned chemical compounds, another important treatment approach is antiviral natural products (NPs) and herbal medicines, which have advantage of safety and effectiveness. These herbal medicines have been used to treat coronavirus infection, including those caused by Middle East respiratory syndrome coronavirus (MERS-CoV), SARS-CoV and SARS-CoV-2 [12–14]. Traditional Chinese medicine (TCM), as a material basis for the application of NPs and herbal medicines under the guidance of theory, have been found to be an effective treatment for COVID-19. Previously, the "three-medicines and three-prescriptions (TMTP)" strategy was recommended as a prescribed formula by the State Council of China because TMTP has exhibited the remarkable therapeutic effects and no side effects, especially against COVID-19 [15]. TMTP mainly include Jinhua Qinggan granules, Lianhua Qingwen granules and capsules, Xuebijing injection, Qingfei Paidu decoction, Huashi Baidu decoction and Xuanfei Baidu decoction, which have presented good clinical efficacy in the treatment of COVID-19 [16]. This strategy halts the progression of the disease and actively alleviates and improves symptoms during the early and middle stages [16–17].
As a combination of several compound medicines, TMTP contains abundant molecules that make the antiviral mechanism unclear. It is necessary to apply an efficient method that quickly and effectively finds the basis of antiviral substances from this valuable natural molecular library. In recent years, the application of in silico technology in drug discovery has achieved prominent success [18–19], supplying sophisticated tools for screening promising lead compounds, predicting potential protein catalytic sites or revealing the mode of protein-ligand interactions. During this pneumonia epidemic, studies have utilized the favored approaches that target SARS-CoV-2 with high-throughput screening of large-scale molecular databases and obtaining potential antiviral drugs [20–21]. With the advancement of computer technology, the combination of computer-aided drug design (CADD) and artificial intelligence (AI) research has become a valuable tool to accelerate the slow process of drug discovery and restraint the expansion of R&D costs, expand the applicable system and improve the level of automation, followed by the development of CADD-based multithreaded in silico screening technology [22–25]. Within the framework of above idea, we proposed a multimodule integrated approach aimed at improving the lead compound screening accuracy and greatly reducing the time cost by fully maximizing the advantages of each module to achieve a semiautomatic pipeline.
Based on the above purposes, an integration of CADD, deep learning (DL) and similarity-based clustering methodologies and experimental validation was adopted in our study as an accurate and efficient means to filter potential anticoronavirus candidates from TMTP database against 3CLpro, the key target of viral replication [26–29]. We expeditiously discovered 5 NPs inhibitors, which further supports that the integrated strategy can accurately and quickly achieve the purpose of obtaining promising lead compounds, suggesting its practicality and worthiness for further optimizing the application form and systems.
Workflow construction and molecular data base constitutions
This integrated screening workflow is divided into four parts (Fig. 1). First, a molecular library was constructed, and the affinity ranking was obtained through molecular docking. Then, cluster analysis was performed to reveal the molecular characteristics of high-affinity clusters and extract the top clusters. Furthermore, combined with high-throughput methods applied to determine the binding affinity and predict the level of activity, the molecules with both excellent properties were finally verified experimentally to determine effective compounds.
In the foremost step, we retrieved 49 Chinese medicinal materials involved in the 6 compound herbal formula in TMTP, and ultimately acquired a total of 3272 NPs to constructed the TMTP molecular library eventually. This library include the Chinese herbal compound prescriptions and the representative Chinese medicines from TMTP as well as the main chemical compositions. On the other hand, we collected 301 of SARS-CoV and 84 of SARS-CoV-2 3CLpro inhibitors, which was performed as comprehensively as possible. The former was used to build DL models, and the latter were treated as a test set. A complete list of the molecules and related information for 3272 TMTP compounds library, compound libraries and SARS-CoV and SARS-CoV-2 3CLpro inhibitors is given in the Supplementary Data 1.
20 clusters divided from the TMTP compound library by cluster analysis
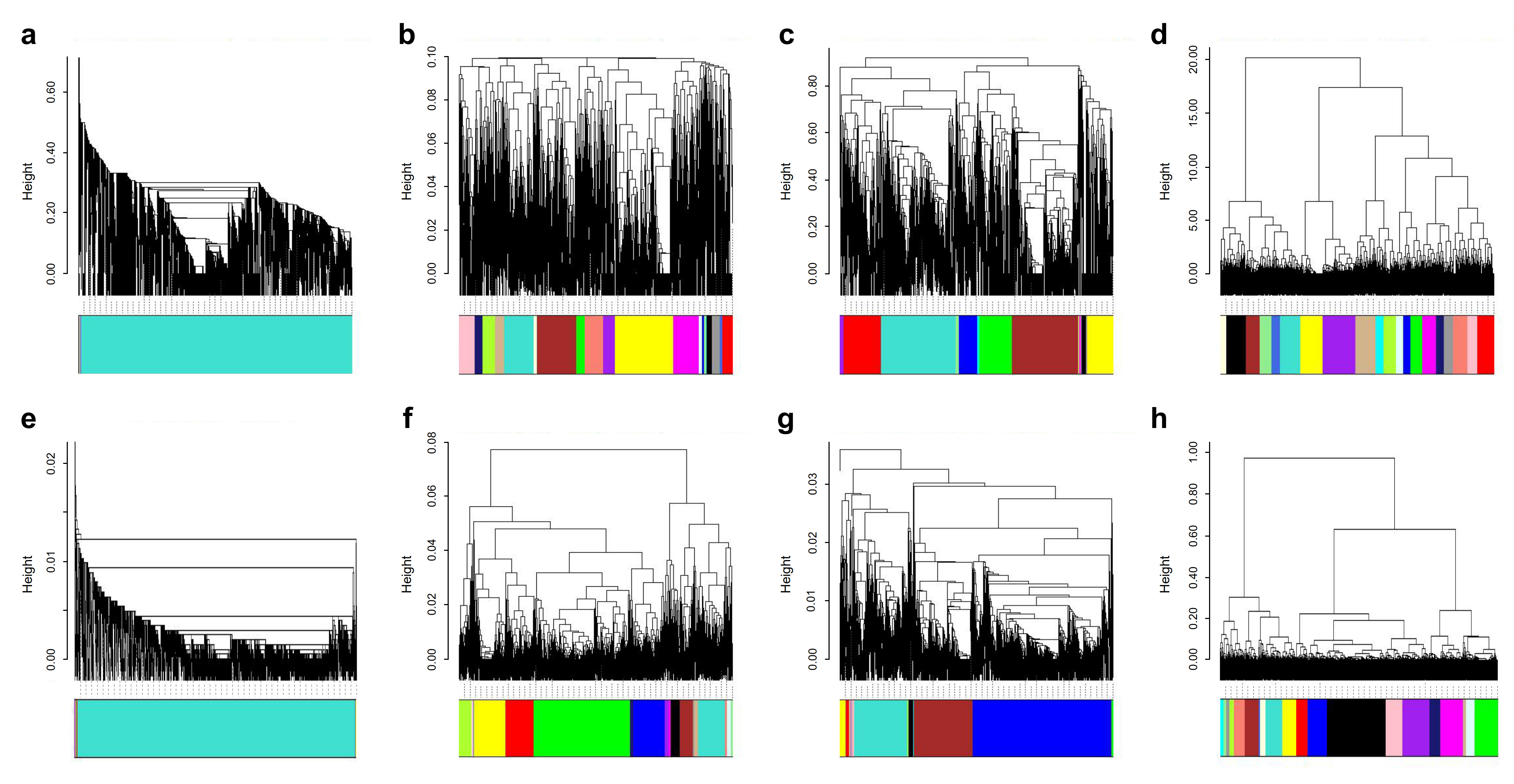
To classify the structural similarity of high-affinity molecules to further narrow the range of lead compounds, a total of 8 combinations between similarities of fingerprint maps and different cluster agglomeration methods were individually used for cluster analysis. Based on the agglomerative coefficient from agnes, we found that the combination of Euclidean and Ward2 exhibited the highest value (agglomerative coefficient = 0.975) compared with that of the other groups, and the agglomerative coefficients of the 8 groups are listed in Supplementary Table 1. Thus, we adopted the Euclidean and Ward2 combination to plot a clustering that contained 20 clusters (k = 20) (Supplementary Fig. 1, Supplementary Table 2).
Dominant clusters determined by means of molecular docking and ECR ranking
Our molecular docking approach was used to obtain the binding ability of the TMTP molecular library with SARS-CoV-2 3CLpro, as well as the affinity score between positive inhibitors with SARS-CoV or SARS-CoV-2 3CLpro for DL modeling. Docking analysis was carried out independently using the programs Autodock Vina, Glide, and MOE. Then, the exponential consensus ranking (ECR) strategy was implemented to reduce the number of false positives. This approach transformed docking scores of a single compound into a decimal number to indicate the comprehensive binding level for the target-ligand complex. Subsequent analyses were performed using ECR values instead of docking scores (Supplementary Data 1).
To compare the binding capacity to 3CLpro among clusters, we calculated and ranked the median, mean and quantile value, etc. of the ECR in each cluster. Then, the dominant clusters were defined as those with a mean ECR value greater than 0.6 (Supplementary Table 3), and 9 dominant clusters were ultimately acquired. Among the 9 dominant clusters, the average ECR value of the royalblue cluster and brown cluster was greater than 0.7, indicating that these two clusters have higher target affinity, while the large number of compounds in the brown cluster suggested that it may have very promising 3CL inhibitors.
Combining binding affinity of SPR and inhibitory activity prediction by DL analysis to narrow the range of hit compounds
In current study, surface plasmon resonance (SPR) was used to rapidly identify molecules in the dominant clusters that have the ability to bind to SARS-CoV-2 3CLpro. As a result, 21 molecules demonstrated high affinity for 3CLpro (Supplementary Table 4). DL was applied in parallel with SPR analysis to predict the 3CLpro inhibitory efficiency of compounds in the dominant clusters and further eliminate the molecules that would be nonspecifically bound in the SPR analysis. As previously described, the collected information on 3CLpro inhibitors (301 SARS-CoV and 84 SARS-CoV-2 inhibitors), including IC50, pIC50, SMILES, and CID, is shown in Supplementary Data 2. After the molecular docking process, we acquired the docking scores and ECR ranking between the SARS-CoV 3CLpro inhibitors and 3CLpro of SARS-CoV and SARS-CoV-2 using three different software programs. Overall, no significant difference between docking scores or ECR of SARS-CoV and SARS-CoV-2 was observed, which was ascribed to the high homology of the SARS-CoV and SARS-CoV-2 3CL proteins [30]. We calculated the similarity index (0.710) of two proteins binding or activity based on the docking matrix. Then, the predicted IC50 of SARS-CoV-2 3CLpro was computed based on the Eq. (1). By means of Rcpi, the molecular descriptors of 301 compounds were extracted as the quantitative structure (Supplementary Data 2). Thereafter, we constructed the quantitative relationship between structure and activity by random forest (RF) and support vector machine (SVM) training classification models. The activity of the 84 compounds for SARS-CoV-2 3CLpro was tested using the training model. We found that the area under curve (AUC) of receiver operating characteristic curve (ROC) in RF was higher than that of SVM (RF: 0.629, SVM: 0.493). Suggesting that the predicted inhibitory value calculated by the RF method was closer to the experimental value than that calculated by the SVM method. Finally, we predicted the activity of 9 dominant clusters; here, a predicted value greater than 0.5 was considered to have an inhibitory effect, and vice versa. A total of 156 compounds were predicted to be active based on statistical analysis. A complete list of the predicted values can be found in Supplementary Data 3.
The molecules that showed affinity with 3CLpro in the SPR analysis and were predicted to be effective via DL were combined and further comprehensively analyzed. Finally, 11 compounds considered promising inhibitor candidates were obtained from the resulting intersection. Interestingly, these high-activity compounds were enriched in the brown, midnightblue and red clusters (Supplementary Table 5). In the above clusters, the brown cluster mainly contains flavonoids and their glycosides. The midnightblue cluster is composed of dammarane and oleanane or their derivative parent nucleus and corresponding glycosides. The compounds in the red cluster are composed of polyhydroxy conjugated systems such as hydroxytyrosol and caffeic acid to connected with sugar units. These types of compounds often exhibit a wide range of biological activities and have also been used in the field of anti-virus [31–32].
5 NPs identifying as potent inhibitors of SARS-CoV-2 3CLpro in vitro
Eleven compounds selected by the virtual screening and DL analysis were subsequently tested using the inhibition assay against SARS-Cov-2 3CLpro. After the initial screening, only five compounds at a concentration of 100 µM demonstrate over 50% inhibitory active against the enzyme. These compounds were able to achieve inhibition at lower concentrations. According to results shown in Fig. 2, narcissoside (MOL003686), kaempferol-3-O-gentiobioside (MOL012143), rutin (MOL000415), vicenin-2 (MOL001543) and isoschaftoside (MOL004958) presented IC50 values of 38.142, 35.892, 31.259, 38.856 and 30.220 µM, respectively. Remarkably, they are all flavonoids. The results of affinity screening by SPR showed that flavonoids accounted for 10 of the 21 compounds and their KD values ranged from 1.525 to 12.46, exhibited strong affinity with 3CLpro (Fig. 3). As demonstrated in Table 1, the KD values are well correlated with IC50 values. Notably, kaempferol-3-o-gentiobioside, vicenin-2 and isoschaftoside were reported as anticoronavirus candidates for the first time due to their inhibition of 3CLpro of SARS-CoV-2. They are very promising for further research to develop compounds with high inhibition efficiency.
| Compound | Molecular Weight (Da) | ECR | KD (µM) | IC50 (µM) |
|---|---|---|---|---|
| Narcissoside | 624.544 | 0.744 | 9.995 | 38.142 |
| Kaempferol-3-O-gentiobioside | 610.518 | 0.861 | 2.694 | 35.892 |
| Rutin | 610.518 | 0.801 | 1.525 | 31.259 |
| Vicenin-2 | 594.518 | 0.771 | 8.583 | 38.856 |
| Isoschaftoside | 564.492 | 0.765 | 11.370 | 30.220 |
Molecular dynamics simulation revealed the stable binding mode of the 5 selected drugs with SARS-CoV-2 3CLpro
The dynamic binding interactions of the five compounds with inhibitory activity were analyzed, and 100 ns molecular dynamics (MD) simulations of ligand-protein complexes were performed. The root mean square deviation (RMSD) of the ligand trajectory was analyzed, revealing that the compound rapidly reached equilibrium within the first 5 ns of the simulation (Fig. 4a), with each value lying between 1.5 and 3.5 Å. Narcissoside and vicenin-2 fluctuated greatly, indicating a flexible bingding to the active site of 3CLpro. In contrast, compounds kaempferol-3-O-gentiobioside, rutin, and isoschaftoside are more fixed.
To explore the binding affinity of each ligand to 3CLpro, the binding free energy was calculated based on MM/PBSA (Table 2). Van der Waals (ΔEvdW) and electrostatic (ΔEele) interactions make major contributions to the binding free energy (Fig. 4e). We observed that rutin exhibited the highest binding affinity to 3CLpro, followed by kaempferol-3-O-gentiobioside, isoschaftoside, vicenin-2 and narcissoside. Analysis of the energy decomposition results of the five compounds suggested that the residues Thr25, Thr26, Ley27, His41, Cys44, Tgr45, Ser46, Met49, Asn142, Gly143, Cys145, His163, His164, Met165, Asp187 and Gln189 mainly contributed to hydrophobic and electrostatic interactions in the 3CLpro-ligand complex (Supplementary Table 6).
| Compound | ΔEvdw | ΔEele | ΔEPB | ΔESA | ΔGTot | ECR |
|---|---|---|---|---|---|---|
| Narcissoside | -58.32 | -21.59 | 73.90 | -6.30 | -16.31 | 0.744 |
| Kaempferol-3-O-gentiobioside | -47.22 | -58.54 | 90.81 | -5.89 | -20.84 | 0.861 |
| Rutin | -45.20 | -43.78 | 78.27 | -5.59 | -22.31 | 0.801 |
| Vicenin-2 | -42.68 | -53.40 | 80.17 | -5.80 | -18.72 | 0.771 |
| Isoschaftoside | -50.61 | -66.58 | 95.47 | -5.95 | -19.67 | 0.765 |
Specifically, from the analysis of the binding interactions, narcissoside showed the least electrostatic interaction (-21.59 kcal/mol), forming hydrogen bonds with Ser46, Gly143, His164, and Glu166. Kaempferol-3-O-gentiobioside forms multiple hydrogen bonds with Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, Glu166, Pro168, and Arg188. Rutin forms hydrogen bond interactions with Thr26, Tyr54, Phe140, Asn142, Gly143, Glu166, and Gln189. Vicenin-2 demonstrated the highest number of H-bonds, forming hydrogen bonds with Thr26, Phe140, Asn142, Gly143 and Glu166. In the analysis of binding energy with isoschaftoside, the contribution of electrostatic interactions to the total binding energy was − 66.58 kJ/mol, which was highest among the 5 compounds, forming H-bond interactions with Thr26, Tyr54, Phe140, Asn142, Gly143 and Glu166, Gln189. The above analysis suggested that flavonoid glycosides provided higher flexibility after forming chains with sugars because of their rotatable bonds, which can bind into pockets and form abundant hydrogen bonds with some key residues. From the perspective of amino acid energy decomposition (Fig. 4d), the compound has a strong interaction with His41, Met49, and Cys145. His41 and Met49 are also the active site residues of 3CLpro [33]. To facilitate the analysis, we first colored the region of the residues His41 and Met49, and then divided the five flavonoids into two categories according to their structural similarity. The active cavity of 3CLpro presented strong hydrophobicity, while the aromatic ring of the flavonoid aglycone provided the main hydrophobic energy contribution in the site. For type A (Fig. 4b), narcissoside, as the only inhibitor with methoxy group. The group has the function of enhancing hydrophobic action of ligand (-58.32 kcal/mol), making the benzene ring easily inserting into the hydrophobic region of the cavity (Fig. 5a, f), resulting in the overall structure extending outside of the cavity and reducing the interaction with residues, eventually reducing the contribution of the binding free energy. In contrast, the flavonoid skeleton of kaempferol-3-O-gentiobioside is close to the cavity (Fig. 5b, g). Furthermore, rutin is inserted into the cavity (Fig. 5c, h), which makes the binding tighter and presents the lowest binding free energy (Table 2). For type B (Fig. 4c), the overall structure shifted in the active pocket due to prolongation of the rigid flavonoid part in vicenin-2 (Fig. 5d, i) and isoschaftoside (Fig. 5e, j), resulting in the distance from the active site being farther than that for kaempferol-3-O-gentiobioside and rutin. However, they did not demonstrate much difference in their total binding free energies.
Notably, from the analysis of the binding interaction, with the key residues, we observed that the interaction strength between His41 and Met49 with the ligand was positively correlated with the affinity of the ligand and 3CLpro binding. In addition, the total energy (Supplementary Table 7) of residues based on the region of His41 to Met49 also exhibited this rule. On the other hand, ΔGTot calculated by MM/PBSA also matched the ECR rank of the molecule (Table 2). The clear binding pattern and significant inhibitory activity of these five flavonoids against 3CLpro indicated that they are promising candidates for anti-SARS-CoV-2 activity. These results prove the correctness of our screening strategy.
To fight the epidemic and obtain effective antiviral drugs from a reservoir of herbal medicines, we designed an integrated pipeline workflow for NP screening. Our integrated strategy combining each submodule into the best workflow and fulfilling the optimization of function therefore exhibited a positive impact from rapid and accurate acquisition of lead compounds to subsequent structural optimization guidance. It is worth noting that the selection and optimization of submodules can be flexibly changed; this integrated strategy may not be limited to NPs screening. Furthermore, this strategy has the potential to derive automated pipelines from prototype workflows to improve the convenience of use while ensuring the accuracy of screening.
The screening pipeline was based on the binding affinity between molecules and targets. To avoid the affinity error caused by limitations of an algorithm of a single software which eliminate compounds that have true binding ability, we selected 3 commonly used docking software programs and converted docking scores into rankings of affinity trends by means of consensus analysis. Attributed to an "or" condition, the ECR assay can reduce the impact of extreme values in software scoring [34], which achieved the fault tolerance of the discrepancy from different scoring functions, that it is very suitable for the research strategy of our study.
When confronted with a large compound library, effective cluster analysis can directly reflect the structural characteristics of molecules or clusters with high affinity and indicate the types of functional groups with high contribution to the corresponding complexes. This helps to guide the subsequent structural optimization and quickly eliminate the set of compounds with poor binding ability to the target. Consequently, similarity-based compound clustering is crucial in pipelines. Among the hierarchical and nonhierarchical clustering methods, Ward's and Jarvis-Patrick are known to be effective algorithms for chemical structure clustering [35–36]. As expected, in the course of practice, Jarvis-Patrick produced too many singletons and a small number of large clusters [37], which is completely inconsistent with our requirements. For the similarity calculation, Tanimoto coefficients and Euclidean distance are the most widely used to evaluate how similar two molecules are to each other [38–39]. Then, we combined them with 4 commonly used hierarchical clustering algorithms and compare the calculated corresponding 8 sets of agglomerative coefficient. Thereafter, we found that Euclidean and Ward2 is the best matches to obtain a cluster of uniform internal structure characteristics, and the number of clusters can also be optimized for specific systems. In summary, this high-precision clustering is suited for but not limited to natural products, and it is worthy of promotion.
As an effective tool for predicting the structure-activity relationship, DL has been widely used in threaded approach [40–41]. The present DL-based activity prediction was capable of describing active molecules from the TMTP database even with a small-volume training set (301 compounds). We calculated the predicted value of SARS-CoV-2 3CLpro from the collected SARS-CoV 3CLpro inhibitory activity according to the similarity matrix between docking data of two target proteins. To build an effective model, we increased molecular character number of the training compound as an independent variable and covered the docking data. Moreover, we compared the predictive accuracy of the SVM and RF methods integrated into the cross validation analysis and found that the AUC value of random forest was higher than SVM, which provided the evidence for the candidates drug discovery by RF-based activity prediction.
Current cluster analysis accurately placed flavonoids into a subset and further obtained the 5 compounds with inhibitory activity of 3CLpro. As compounds characterized by 2-phenyl-benzyl-γ-pyrone nucleus, flavonoids are particularly valuable NPs that possess anti-inflammatory [42], antioxidant [43], anti-microbial [44], and even antiviral activities. In a study of inhibitors against coronavirus, rhoifolin, pectolinarin, herbacetin [45] and amentoflavone [46] were demonstrated to block the function of SARS-CoV 3CLpro. The glycoside derivatives of kaempferol also proved to be virus release inhibit agents by blocking the 3a channel [47]. During the COVID-19 outbreak, narcissoside [48] and rutin [29] also exhibited the inhibitory effects on main protease of SARS-CoV-2, which is confirmed the practicality of our screening strategy.
The limitations of this study have been presented. First, on account of the few reports of tested compounds, a relatively small sample training set in the DL analysis was available. When more positive drugs are involved, the accuracy of DL activity prediction can be further improved. Second, the 5 NPs have not been verified to have inhibitory activity by in vivo experimental and preclinical data due to the powerful invasion ability of SARS-CoV-2.
In conclusion, this study successfully employed an integrated screening strategy to identify 5 potential inhibitors of SARS-CoV-2 3CLpro from a NPs library composed of clinically effective herbal medicines. On the basis of this research, further research is worth pursuing to produce derivatives that can produce better inhibitors. The high efficiency and accurate characteristics of this strategy greatly shorten the hit cycle of lead compounds in the process of drug discovery for acute diseases and accelerate the process of drug development. We recommend that this integrated screening strategy be applied to other targets that urgently need effective drugs.
Construction of TMTP Chemical Constituent Databases
All chemical ingredients from 49 crude Chinese Medicine were extracted from the TCMSP database (Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform, http://lsp.nwu.edu.cn/tcmsp.php). We stringently corrected the detailed information of total ingredients from the literature mining and different TCM-related databases to ensure the accuracy and completeness of the library. Then, the OpenBabel toolkit was used to convert the mol2 file of each molecule into unified SDF, pdbqt and SMILES file formats to prepare for molecular docking. Subsequently, we retrieved the absorption, distribution, metabolism and excretion (ADME) properties data from TCMSP database, containing molecular weight (MW), oral bioavailability (OB), number of hydrogen-bond acceptors (HBA), number of hydrogen-bond donors (HBD), etc. Other molecular descriptors, including atom additive logP (ALogP), atom molar refractivity values (AMR), and topological polar surface area (TPSA), were calculated based on the Rcpi in the R platform.
Molecular docking
To accurately predict docking poses, three different molecular docking programs, AutoDock Vina [49] (version 1.1.2), Maestro (version 11.4, Schrödinger, LLC, New York, NY, 2021), and molecular operating environment software (MOE, Chemical Computing Group, version 2019.0101), were used to detect the binding capability between diverse compounds and SARS-CoV or SARS-CoV-2 3CLpro. The protein and ligand were prepared for the docking process. First, the crystal structural file of SARS-CoV and SARS-CoV-2 3CLpro were downloaded from protein database (PDB ID 3V3M [50], 6LU7 [51]). For the target protein, the preparation included carrying out the correction of chemical bond sequence, hydrogenation, treatment of disulfide bonds, metal ions, and removal of water molecules, heteroatoms with default settings in three software. For ligand preparation, apart from AutoDock Vina which minimized the compound’s energy in Chem3D software, Maestro and MOE were carried out with inner LigPrep and Energy Minimize protocol respectively, to generated the correct form, and all the hydrogen atoms and the torsion information were added. To ensure the uniformity of different software at the docking position, the binding site box of 3CLpro coincided with binding site of the original inhibitors. Docking analysis was conducted with default protocol in AutoDock Vina, the Extra precision (XP) was used in GlideScore scoring functions [52], and the Induced Fit module was chosen in MOE. The PyMOL Molecular Graphics System (version 2.0, Schrödinger, LLC) was used to visualize the docking posture of compounds at the binding pocket of 3CLpro.
Consensus analysis
To combine results from several docking programs, we adopted ECR methods proposed by Karen Palacio-Rodríguez et al. [34]. In brief, ECR methods assigned an exponential score to each molecule for each scoring function based on the rank of the molecules provided by different docking program. As shown in equation (2), represented the expected value of an exponential distribution and was assigned to be 10. The ultimate ECR score of each molecule was defined as the sum of all the exponential scores , which was a positive correlation between the ECR value and affinity of compound against target protein.

Cluster analysis
To identify homogeneous and distinct groups, or similar objects in TMTP NPs datasets, we performed clustering analysis with the ChemmineR and WGCNA packages. First, we collected the SDF file of each molecule produced by the OpenBabel software. By means of readMolFromSDF, we converted the SDF files into mol files. The SDF files were loaded into the ChemmineR for calculating atom pair fingerprints (APfp) of all compounds, which were used for calculate the structural similarity between the different compounds [53-54]. We adopted the Tanimoto coefficients and Euclidean distance for computing distance or dissimilarity metrics based on the fingerprint of TMTP natural products. To classify the different compounds into a series of the relative number of clusters, we concentrated on single, complete, average, and Ward's algorithms to map the strength of the clustering results. Additionally, the dendrograms of 8 hclust approaches were plotted, and the agnes from dendextend computing the agglomerative coefficient was used to measure the amount of clustering structure found (values closer to 1 suggest a strong clustering structure). To clearly delineate the different clusters, we integrated the WGCNA, which can effectively assign different modules and are represented as visualized colors. The functions of cutreeDynamic and labels2colors were further used to investigate the best clustering results.
Surface plasmon resonance analysis
The binding studies were performed at 25 ℃ on a Biacore T200 instrument at a flow rate of 30 μL/min in running buffer composed of PBS (pH 7.4) and 3 mM EDTA. CM7 chips activated in a 10 min injection procedure with a mixture of EDC (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide)/NHS (Nhydroxysuccinimide) (0.2 M/0.05 M) and immobilized with anti-histone antibody until levels of immobilization were between 15000 and 20000 RU. The chip was then deactivated with a 7 min injection of 1 M ethanolamine (pH 8.0). The 3CLpro-his protein was then applied on the chip to reach typically levels between 4000 and 5000 RU. The binding activity and stability of proteins to ligands were tested at the end of each compound injection. The association and dissociation phases of tested ligands were monitored for 120 s each. The final binding experiment with small molecule ligands was performed in the above running buffer supplemented with 5% DMSO. PBS, EDTA, CM7 chip, EDC, NHS and ethanolamine were obtained from GE, 3CLpro-his was obtained from Kangma- Healthcodea.
Deep learning analysis
3CLpro Positive inhibitors of SARS-CoV and SARS-CoV-2 for individual modeling and validation via DL were acquired from the literature, related IC50 were collected, and the SDF files was downloaded from Pubchem or generated by ChemDraw, as well as SMILES files. Then, each 3CLpro inhibitor of SARS-CoV was docked with the 3CLpro of SARS-CoV and SARS-CoV-2 separately, as described in the docking procedure above, ECR ranking was constructed from the docking score matrix. The inhibitory efficiency in the data set was converted to two classes: active (pIC50 ≥ 6) and inactive (pIC50 < 6). Then, the 3CLpro similarity index (SI) between SARS-CoV and SARS-CoV-2 was calculated via SMI from MatrixCorrelation [55]. To build the predicted model, we proposed the equation (1) to transform the SARS-CoV IC50 to predicted IC50 of the SARS-CoV-2 inhibition efficiency. Subsequently, a regression model based on the quantitative structure and activity relationship of SARS-CoV-2 was constructed.

The inhibitory compounds targeting SARS-CoV-2 3CLpro and the related IC50 were validated as a test data set. By means of Rcpi, we extracted the molecular descriptors of the validated compounds. The molecular descriptors and docking value of the training data sets were input as independent variables, and the predicted IC50 values were input as dependent variables. We selected the random forest (RF) and support vector machine (SVM), which are widely used DL methods [56-57]. The accuracy of two classification model was evaluated by the AUC value of ROC. Based on the AUC value, we selected the optimal methods to predict the inhibition efficiency of natural products from dominant clusters.
SARS-CoV-2 3CLpro inhibition assay
The inhibition assay of SARS-CoV-2 3CLpro was carried out based on the reported method [58]. In the initial step, 0.5 µg of SARS-CoV-2 3CLpro was preincubated with 100 µL of 200 µM test compound at room temperature for 15 minutes. Then, the reaction was triggered after the addition of 10 µM Dabcyl-KTSAVLQSGFRKME-Edans (GL Biochem). The fluctuation of fluorescence intensity was monitored on a GENios microplate reader (Tecan), where the excitation wavelength was 340 nm and the emission wavelength was set to 490 nm. Control reactions were performed under the same condition, but the compounds or enzymes were excluded from the reaction system. The median inhibitory concentration (IC50) values against SARS-CoV-2 3CLpro was calculated by nonlinear regression analysis via GraphPad Prism 7.03 (GraphPad Software, San Diego, CA, USA).
Molecular dynamics simulation and binding free energy calculation
MD simulations of the screened natural inhibitors of 3CLpro were performed with Amber14 [59] to evaluate their binding interaction patterns with 3CLpro. The protein-ligand complexes were used as the initial structure for subsequent MD simulations. The ligand and protein were treated with General Amber Force Field (GAFF [60]) and FF14SB [61] respectively. For the amino acid residues of the protein, the default protonation state in Amber14 was adopted, and the hydroprocessing was carried out using tleap module. By means of the Gaussian09 software package [62], the inhibitor molecules optimization and the electrostatic potential calculation based on B3LYP/6-31G. The partial charges of small molecules are obtained by the electrostatic potential fitting of the Antechamber module using the RESP method.
The Gaussian09 software package was dedicated to optimize the structure of all inhibitor molecules and calculate the electrostatic potential based on B3LYP/6-31G. The partial charges of small molecules were obtained by the electrostatic potential fitting of the Antechamber module. After adding counter ions to each complex to maintain the neutrality of the system, the entire system was contained to a TIP3P rectangular water box. Furthermore, energy minimization was performed by steepest descent method of 2500 steps and conjugate gradient method of 2500 steps. Subsequently, same methods were used to optimize the unconstrained system. The Particle mesh Ewald (PME) was performed in the MD simulation to deal with the long-range electrostatic interaction, and the SHAKE algorithm was used to constrain all the bonds connected to the hydrogen atom and the time step was set to 2 fs. Then set a cutoff value of 10 Å for non-bonding interactions. The constrained whole system was heated from 0 K to 300 K in 60 ps at a constant volume, subsequently, the solvent density was balanced in a constant pressure and thermostatic system (T = 300 K, P = 1 ATM). Followed by 100 ns of MD simulations control at constant pressure. Save one frame per ps for subsequent analysis at last.
COVID-19: severe situation of coronavirus disease 2019; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; 3CLpro: 3-chymotrypsin-like protease; NPs: natural products; MERS-CoV: Middle East respiratory syndrome coronavirus; TCM: Traditional Chinese medicine; TMTP: three-medicines and three-prescriptions; CADD: combination of computer-aided drug design; AI: artificial intelligence; DL: deep learning; ECR: exponential consensus ranking; SPR: surface plasmon resonance; RF: random forest; SVM: support vector machine; AUC: area under curve; ROC: receiver operating characteristic curve; MD: molecular dynamics; RMSD: root mean square deviation; ADME: absorption, distribution, metabolism and excretion; MW: molecular weight; OB: oral bioavailability; HBA: hydrogen-bond acceptors; HBD: number of hydrogen-bond donors; AlogP: atom additive logP; AMR: atom molar refractivity values; TPSA: topological polar surface area; ApfP: atom pair fingerprints.
Availability of data and materials
The authors declare that all data and code supporting the findings of this study are available within the paper and its supplementary information files.
Competing interests
The authors declare no competing interests.
Funding
This work was supported by National Natural Science Foundation of China (31871281) , Shanghai Municipal Health Commission (2020XGKY12) and Scientific Research Foundation for Advanced Talents of Shanghai University of Traditional Chinese Medicine.
Authors’ contributions
Zhengtao Wang, Li Yang and Qingzhong Wang designed the project. Material preparation and data collection and analysis were performed by Qi Liao and Ziyu Chen. Yanlin Tao and Beibei Zhang measured SPR and inhibition assay. The first draft of the manuscript was written by Qi Liao and Ziyu Chen. Qingzhong Wang and Li Yang checked and edited the manuscripts. All authors read and approved the final version of the manuscript.
Acknowledgements
The authors would like to thank Dr. Guixin Chou and Dr. Lili Ji for their helpful advice and comments.
- Grant MC, Geoghegan L, Arbyn M, et al (2020) The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): A systematic review and meta-analysis of 148 studies from 9 countries. PLoS One 15:e0234765. doi:10.1371/journal.pone.0234765.
- Dong E, Du H, Gardner L (2020) An interactive web-based dashboard to track COVID-19 in real time. Lancet. Infect. Dis. 20:533-534. doi:10.1016/s1473-3099(20)30120-1.
- Rosa SGV, Santos WC (2020) Clinical trials on drug repositioning for COVID-19 treatment. Rev. Panam. Salud. Publica. 44:e40. doi:10.26633/RPSP.2020.40.
- de Vries M, Mohamed AS, Prescott RA, et al (2020) Comparative study of a 3CL pro inhibitor and remdesivir against both major SARS-CoV-2 clades in human airway models. bioRxiv: 2020.08.28.272880. https://doi.org/10.1101/2020.08.28.272880.
- Gordon CJ, Tchesnokov EP, Woolner E, et al (2020) Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 295:6785-6797. doi:10.1074/jbc.RA120.013679.
- Vuong W, Khan MB, Fischer C, et al (2020) Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 11:4282. doi:10.1038/s41467-020-18096-2.
- Riva L, Yuan S, Yin X, et al (2020) Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586:113-119. doi:10.1038/s41586-020-2577-1.
- Yamamoto N, Matsuyama S, Hoshino T, Yamamoto N (2020) Nelfinavir inhibits replication of severe acute respiratory syndrome coronavirus 2 in vitro. bioRxiv: 2020.04.06.026476. https://doi.org/10.1101/2020.04.06.026476.
- Salas Rojas M, Silva Garcia R, Bini E, et al (2021) Quinacrine, an Antimalarial Drug with Strong Activity Inhibiting SARS-CoV-2 Viral Replication In Vitro. Viruses 13:121. doi:10.3390/v13010121.
- Beigel JH, Tomashek KM, Dodd LE, et al (2020) Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 383:1813-1826. doi:10.1056/NEJMoa2007764.
- Joshi S, Parkar J, Ansari A, et al (2021) Role of favipiravir in the treatment of COVID-19. Int. J. Infect. Dis. 102:501-508. doi:10.1016/j.ijid.2020.10.069.
- Lin SC, Ho CT, Chuo WH, et al (2017) Effective inhibition of MERS-CoV infection by resveratrol. BMC Infect. Dis. 17:144. doi:10.1186/s12879-017-2253-8.
- Leung PC (2007) The efficacy of Chinese medicine for SARS: a review of Chinese publications after the crisis. Am. J. Chin. Med. 35:575-581. doi:10.1142/S0192415X07005077.
- Shahrajabian MH, Sun W, Shen H, Cheng Q (2020) Chinese herbal medicine for SARS and SARS-CoV-2 treatment and prevention, encouraging using herbal medicine for COVID-19 outbreak. Acta Agric. Scand., Sect. B 70:437-443. doi:10.1080/09064710.2020.1763448.
- The state council information office, Press Conference of the Joint Prevention and Control Mechanism, http://www.gov.cn/xinwen/gwylflkjz92/index.htm. Accessed 14 Apr 2020.
- Huang YF, Bai C, He F, et al (2020) Review on the potential action mechanisms of Chinese medicines in treating Coronavirus Disease 2019 (COVID-19). Pharmacol. Res. 158:104939. doi:10.1016/j.phrs.2020.104939.
- Hu K, Guan Wj, Bi Y, et al (2020) Efficacy and safety of Lianhuaqingwen capsules, a repurposed Chinese herb, in patients with coronavirus disease 2019: A multicenter, prospective, randomized controlled trial. Phytomedicine. doi:10.1016/j.phymed.2020.153242.
- Reynolds CH (2014) Impact of computational structure-based methods on drug discovery. Curr. Pharm. Des. 20:3380-3386. doi:10.2174/138161282020140528105532.
- Nicolaou CA, Humblet C, Hu H, et al (2019) Idea2Data: Toward a New Paradigm for Drug Discovery. ACS Med. Chem. Lett. 10:278-286. doi:10.1021/acsmedchemlett.8b00488.
- Fischer A, Sellner M, Neranjan S, et al (2020) Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 21:3626. doi:10.3390/ijms21103626.
- Elmezayen AD, Al-Obaidi A, Şahin AT, Yelekçi K (2020) Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. doi:10.1080/07391102.2020.1758791.
- Gupta V, Crudu A, Matsuoka Y, et al (2019) Multi-dimensional computational pipeline for large-scale deep screening of compound effect assessment: an in silico case study on ageing-related compounds. npj Syst. Biol. Appl. 5:42. doi:10.1038/s41540-019-0119-y.
- Samdani A, Vetrivel U (2018) POAP: A GNU parallel based multithreaded pipeline of open babel and AutoDock suite for boosted high throughput virtual screening. Comput. Biol. Chem. 74:39-48. doi:10.1016/j.compbiolchem.2018.02.012.
- Harnie D, Saey M, Vapirev AE, et al (2017) Scaling machine learning for target prediction in drug discovery using Apache Spark. Future Gener. Comput. Syst. 67:409-417. doi:10.1016/j.future.2016.04.023.
- Krivák R, Hoksza D (2018) P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure. J. Cheminf. 10:39. doi:10.1186/s13321-018-0285-8.
- Ziebuhr J, Snijder EJ, Gorbalenya AE (2000) Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 81:853-879. doi:10.1099/0022-1317-81-4-853.
- Kumar V, Tan KP, Wang YM, et al (2016) Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 24:3035-3042. doi:10.1016/j.bmc.2016.05.013.
- Wang L, Bao BB, Song GQ, et al (2017) Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: Chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem. 137:450-461. doi:10.1016/j.ejmech.2017.05.045.
- Jo S, Kim S, Kim DY, et al (2020) Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzyme. Inhib. Med. Chem. 35:1539-1544. doi:10.1080/14756366.2020.1801672.
- Tahir Ul Qamar M, Alqahtani SM, Alamri MA, Chen LL (2020) Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 10:313-319. doi:10.1016/j.jpha.2020.03.009.
- Song X, Yuan G, Li P, Cao S (2019) Guanidine-Containing Polyhydroxyl Macrolides: Chemistry, Biology, and Structure-Activity Relationship. Molecules 24:3913. doi:10.3390/molecules24213913.
- Padhye S, Chavan D, Pandey S, et al (2010) Perspectives on Chemopreventive and Therapeutic Potential of Curcumin Analogs in Medicinal Chemistry. Mini-Rev. Med. Chem. 10:372-387. doi:10.2174/138955710791330891.
- Gul S, Ozcan O, Asar S, et al (2020) In silico identification of widely used and well-tolerated drugs as potential SARS-CoV-2 3C-like protease and viral RNA-dependent RNA polymerase inhibitors for direct use in clinical trials. J. Biomol. Struct. Dyn. doi:10.1080/07391102.2020.1802346.
- Palacio-Rodríguez K, Lans I, Cavasotto CN, Cossio P (2019) Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 9:5142. doi:10.1038/s41598-019-41594-3.
- Varin T, Bureau R, Mueller C, Willett P (2009) Clustering files of chemical structures using the Szekely-Rizzo generalization of Ward's method. J. Mol. Graphics Modell. 28:187-195. doi:10.1016/j.jmgm.2009.06.006.
- Malhat MG, Mousa HM, El-Sisi AB, (2014) Improving Jarvis-Patrick algorithm for drug discovery. 2014 9th International Conference on Informatics and Systems. Cairo, Egypt, 2014.
- Khanna V, Ranganathan S (2010) Molecular similarity and diversity approaches in chemoinformatics. Drug Dev. Res. 72:74-84. doi:10.1002/ddr.20404.
- Raymond JW, Blankley CJ, Willett P (2003) Comparison of chemical clustering methods using graph- and fingerprint-based similarity measures. J. Mol. Graphics Modell. 21:421-433. doi:10.1016/S1093-3263(02)00188-2.
- Downs GM, Willett P, Fisanick W (1994) Similarity Searching and Clustering of Chemical-Structure Databases Using Molecular Property Data. J. Chem. Inform. Comput. Sci. 34:1094-1102. doi:10.1021/ci00021a011.
- Dey S, Luo H, Fokoue A, et al (2018) Predicting adverse drug reactions through interpretable deep learning framework. BMC Bioinf. 19:476. doi:10.1186/s12859-018-2544-0.
- Mayr A, Klambauer G, Unterthiner T, Hochreiter S (2016) DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 3:80. doi:10.3389/fenvs.2015.00080.
- Serafini M, Peluso I, Raguzzini A (2010) Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 69:273-278. doi:10.1017/s002966511000162x.
- Pietta P-G (2000) Flavonoids as Antioxidants. J. Nat. Prod. 63:1035-1042. doi:10.1021/np9904509.
- Xie Y, Yang W, Tang F, et al (2014) Antibacterial Activities of Flavonoids: Structure-Activity Relationship and Mechanism. Curr. Med. Chem. 22:132-149. doi:10.2174/0929867321666140916113443.
- Jo S, Kim S, Shin DH, Kim MS (2020) Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzyme. Inhib. Med. Chem. 35:145-151. doi:10.1080/14756366.2019.1690480.
- Ryu YB, Jeong HJ, Kim JH, et al (2010) Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg. Med. Chem. 18:7940-7947. doi:10.1016/j.bmc.2010.09.035.
- Schwarz S, Sauter D, Wang K, et al (2014) Kaempferol Derivatives as Antiviral Drugs against the 3a Channel Protein of Coronavirus. Planta Med. 80:177-182. doi:10.1055/s-0033-1360277.
- Dubey K, Dubey R (2020) Computation screening of narcissoside a glycosyloxyflavone for potential novel coronavirus 2019 (COVID-19) inhibitor. Biomed. J. 43:363-367. doi:10.1016/j.bj.2020.05.002.
- Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31:455-461. doi:10.1002/jcc.21334.
- Jacobs J, Grum-Tokars V, Zhou Y, et al (2013) Discovery, synthesis, and structure-based optimization of a series of N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 56:534-546. doi:10.1021/jm301580n.
- Jin Z, Du X, Xu Y, et al (2020) Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 582:289-293. doi:10.1038/s41586-020-2223-y.
- Friesner RA, Murphy RB, Repasky MP, et al (2006) Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 49:6177-6196. doi:10.1021/jm051256o.
- Bajusz D, Racz A, Heberger K (2015) Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminf. 7:20. doi:10.1186/s13321-015-0069-3.
- O'Boyle NM, Sayle RA (2016) Comparing structural fingerprints using a literature-based similarity benchmark. J. Cheminf. 8:36. doi:10.1186/s13321-016-0148-0.
- Indahl UG, Næs T, Liland KH (2018) A similarity index for comparing coupled matrices. J. Chemom. 32:e3049. doi:10.1002/cem.3049.
- Manavalan B, Shin TH, Lee G (2018) DHSpred: support-vector-machine-based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 9:1944-1956. doi:10.18632/oncotarget.23099.
- Inglada J, Vincent A, Arias M, Marais-Sicre C (2016) Improved Early Crop Type Identification By Joint Use of High Temporal Resolution SAR And Optical Image Time Series. Remote Sensing 8:362. doi:10.3390/rs8050362.
- Chen L, Gui C, Luo X, et al (2005) Cinanserin Is an Inhibitor of the 3C-Like Proteinase of Severe Acute Respiratory Syndrome Coronavirus and Strongly Reduces Virus Replication In Vitro. J. Virol. 79:7095-7103. doi:10.1128/jvi.79.11.7095-7103.2005.
- Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III,Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA (2014), AMBER 14, University of California, San Francisco.
- Wang J, Wolf RM, Caldwell JW, et al (2004) Development and testing of a general amber force field. J. Comput. Chem. 25:1157-1174. doi:10.1002/jcc.20035.
- Maier JA, Martinez C, Kasavajhala K, et al (2015) ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 11:3696-3713. doi:10.1021/acs.jctc.5b00255.
- Gaussian 09, Revision D.01, Frisch MJ, Trucks GW, Schlegel HB,. Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JE, Bakken V, Adamo V, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi AJ, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ, Gaussian, Inc., Wallingford CT, 2013.
- FigureS100.png
The dendrogram of the 8 combination between similarities of fingerprint maps and different cluster agglomeration. a Group of tanimoto and single. b Group of tanimoto and complete. c Group of tanimoto and average. d Group of tanimoto and ward2. e Group of euclidean and single. f Group of euclidean and complete. g Group of euclidean and average. h Group of euclidean and ward2.
- SupplementaryData1.xlsx
The compound in.formation was summarized in the tables including TMTP molecular library, DL training set (SARS-CoV 3CLpro inhibitors) and test set (SARS-CoV-2 3CLpro inhibitors). In addition, the docking scores of TMTP library compounds with SARS-CoV-2 3CLpro, training set compounds with SARS-CoV and SARS-CoV-2 3CLpro and test set compounds with SARS-CoV-2 3CLpro, the corresponding ECR ranking scores were also included.
- SupplementaryData2.xlsx
In the excel tables, training set and test set molecular descriptors were calculated for DL modeling and optimization.
- SupplementaryData3.xlsx
The compound activity data predicted by DL. Compounds of test sets and dominant clusters were analyzed respectively through DL to predict their inhibitory activity against SARS-CoV-2 3CLpro. The value of “Pred. act.” is proportional to the intensity of the potential inhibitory activity, and it is considered as no inhibitory activity if it is less than 0.5.
{kind=link}