General methods
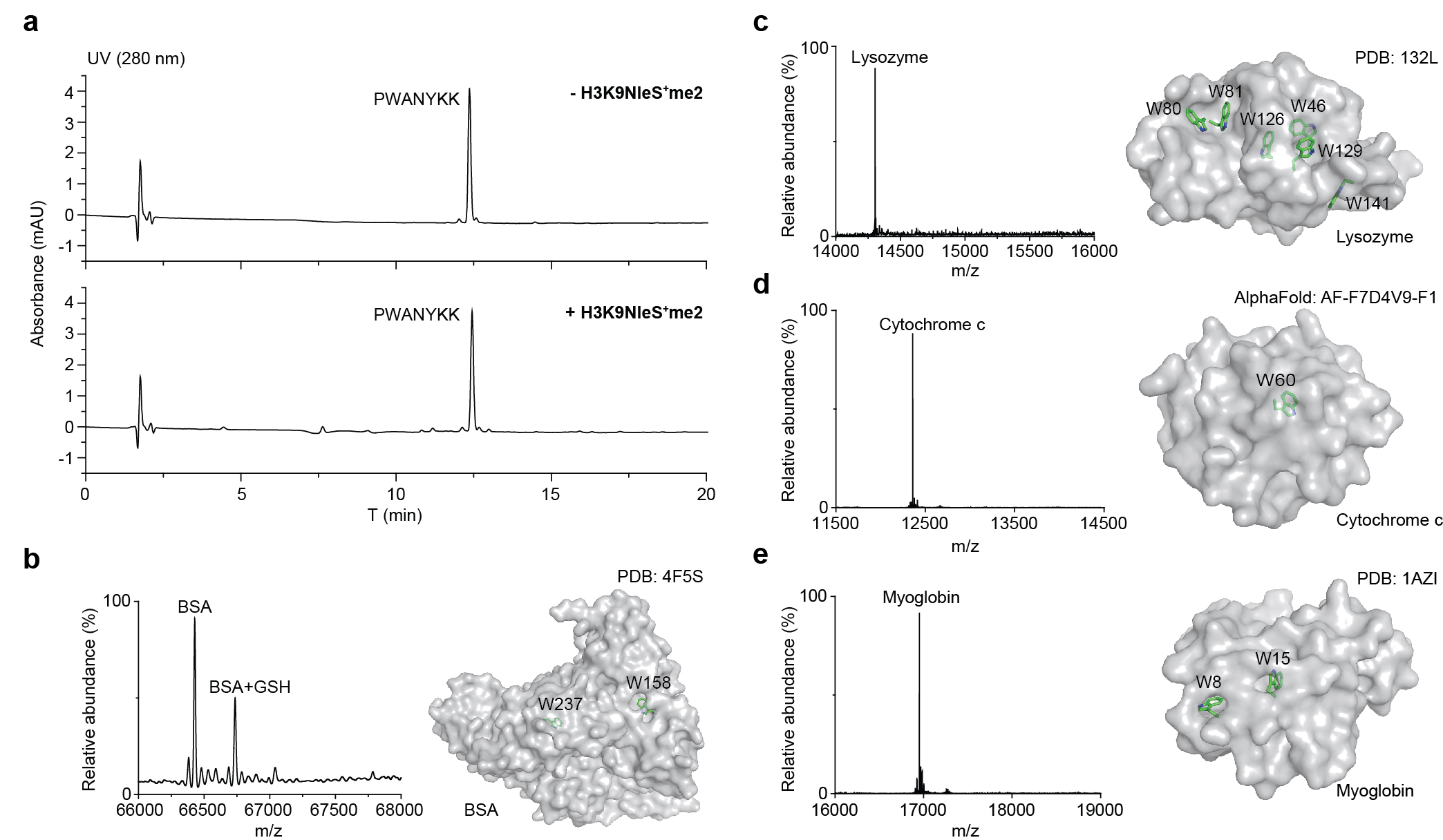
All reagents and chemicals were purchased from Energy Chemical, Macklin, and Bidepharmatech, and used as received without any further purification. Bis-PEG5-NHS ester was purchased from Leyan. All Fmoc-protected amino acids, rink amide resin, and Fmoc-HoMet-OH were purchased from GL Biochem Co., Ltd. (China). Some peptides were purchased from Bankpeptide Biological Technology Co., Ltd (China) including histone methyllysine peptides for sulfonium binding competition, N-FITC (fluorescein isothiocyanate) histone methyllysine peptides for fluorescence polarization assay, H3K4M and H3K9M for Met+me peptide preparation, and histone peptides with biotin for chemical crosslinker assay. NMR spectra were recorded on 500 MHz or 600 MHz Bruker BioSpin, Switzerland. UPLC-Q-TOF-MS analyses were performed with G2 XS high resolution mass spectrometer using Waters Acquity UPLC BEH C18 (1.7 µm, 2.1 × 50 mm) or Waters Acquity UPLC Protein BEH C4 (1.7 µm, 2.1 × 50 mm). Linear gradients using A: H2O (0.1% HCOOH) and B: CH3CN (0.1% HCOOH) over varying periods of time. Bruker rapiflex MALDI-TOF-MS was used for characterization of peptides. Semi-preparative HPLC was carried out on a Waters 1525 pump with 2489 detector using a XBrigde BEH C18 (10 µm, 19 × 250 mm) column. Linear gradients using A: H2O (0.1% TFA) and B: CH3CN (0.1% TFA) over varying periods of time. Peptide centrifugation was performed by highspeed refrigerated micro centrifuge MX-307 purchased from TOMY KOGYO Co., Ltd. Peptide freeze drying was achieved by Labconco FreeZone Benchtop Freeze Dryer. 302 nm or 365 nm light source was performed on a Analytikjena UVP Crosslinker CL-1000. SCHOTT N-WG305 50x50mm 1mm T LP Filter (14466) was purchased from Edmund Optics. BSA (A8020) was purchased from Solarbio. Lysozyme (L6876), myoglolin (M0630), and cytochrome c (C7752) were purchased from Sigma-Aldrich.
Chemical synthesis methods
The detailed synthetic methods, NMR spectra and mass spectra for all the peptides and small molecules are provided in the Supplementary Information.
Recombinant protein expression and purification
The chromodomains from human CBX1 (residues 20-73, C60A) and human MPP8 (residues 55-116) were cloned into pET-21(+) vector with a C-terminal 6×His tag. Proteins were over-expressed in BL21(DE3) Escherichia coli cells by induction of 0.25 mM isopropyl β-D-thiogalactoside at 30 ºC for 3 h when OD600 reached 0.6-0.8 in the LB medium. Harvested cells were suspended in lysis buffer (20 mM Tris, 150 mM NaCl, 0.2 mM PMSF, pH 7.5) and then lysed by sonication. The clarified lysate by centrifugation was applied to nickel resin equilibrated in lysis buffer. The resin was washed sequentially by lysis buffer, high salt buffer (20 mM Tris, 500 mM NaCl, pH 7.5) and 20 mM imidazole buffer (20 mM Tris, 150 mM NaCl, 20 mM imidazole, pH 7.5). The target proteins were eluted by 200 mM imidazole buffer (20 mM Tris, 150 mM NaCl, 200 mM imidazole, pH 7.5). Purified proteins in the storage buffer (20 mM Tris, 150 mM NaCl, pH 7.5) were finally obtained after dialysis and concentration. The proteins were snap-frozen and stored at -80 ºC.
The detailed expression and purification procedure of the other recombinant proteins in this study are provided in the Supplementary Information
Isothermal titration calorimetry
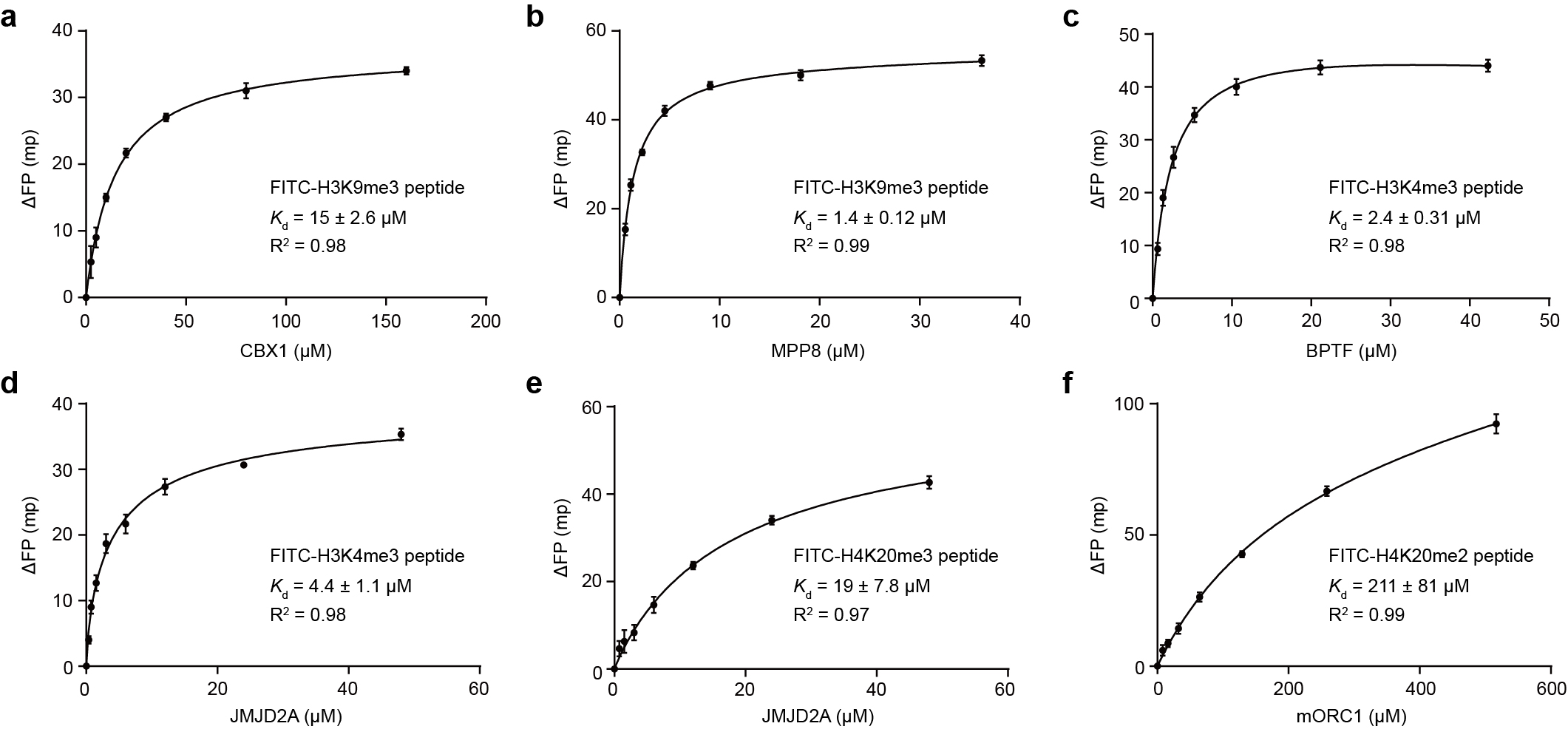
ITC experiments were conducted at 25 ºC using MicroCal PEAQ-ITC Automated (Malvern Instruments) by titrating peptides into proteins at 25 ºC. To the chromo domain of CBX1 in 20 mM Tris, 150 mM NaCl, pH 7.5, H3(1-15) peptides containing mono-, di- or tri methylation at K9 and H3K9NleS+me2 peptide (S16) (250-1000 μM) in the same buffer were titrated into proteins at 25 μM. A total of 19 injections were performed with 0.4 μL for the first and 2.0 μL for the rest. Each spacing was 150 s and the reference power was 10 μcal/s. Data was modeled using the “One Set of Sites” supplied in MicroCal PEAQ-ITC Analysis software (version 1.30). The resultant ITC curves were processed using GraphPad Prism software.
Sulfonium-mediated crosslinking to methyllysine readers
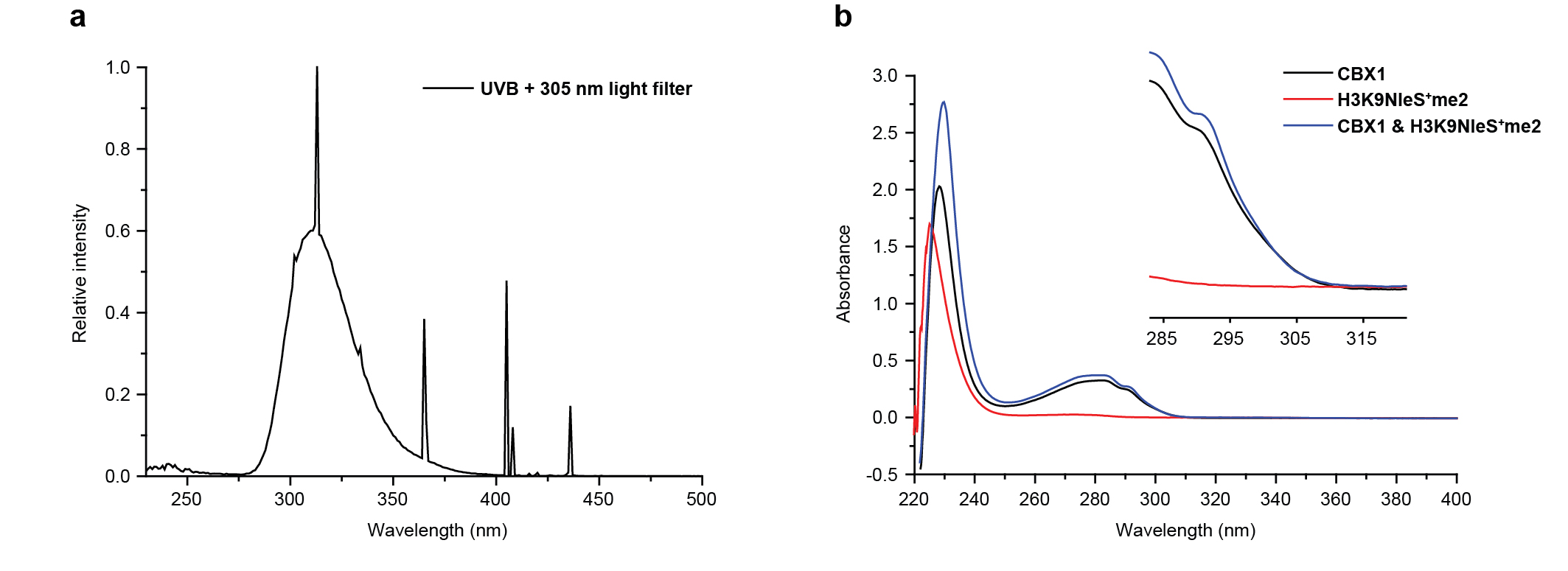
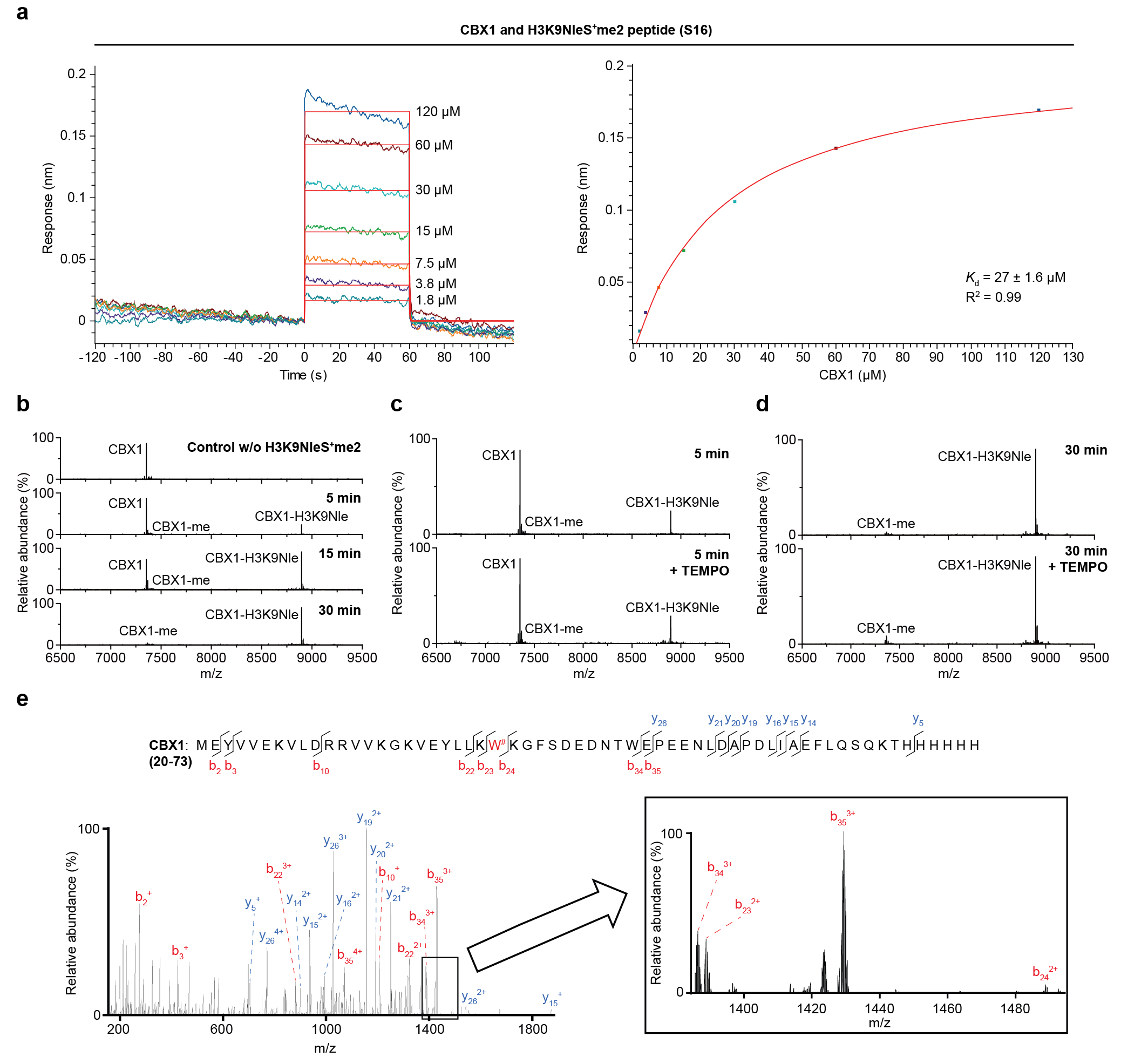
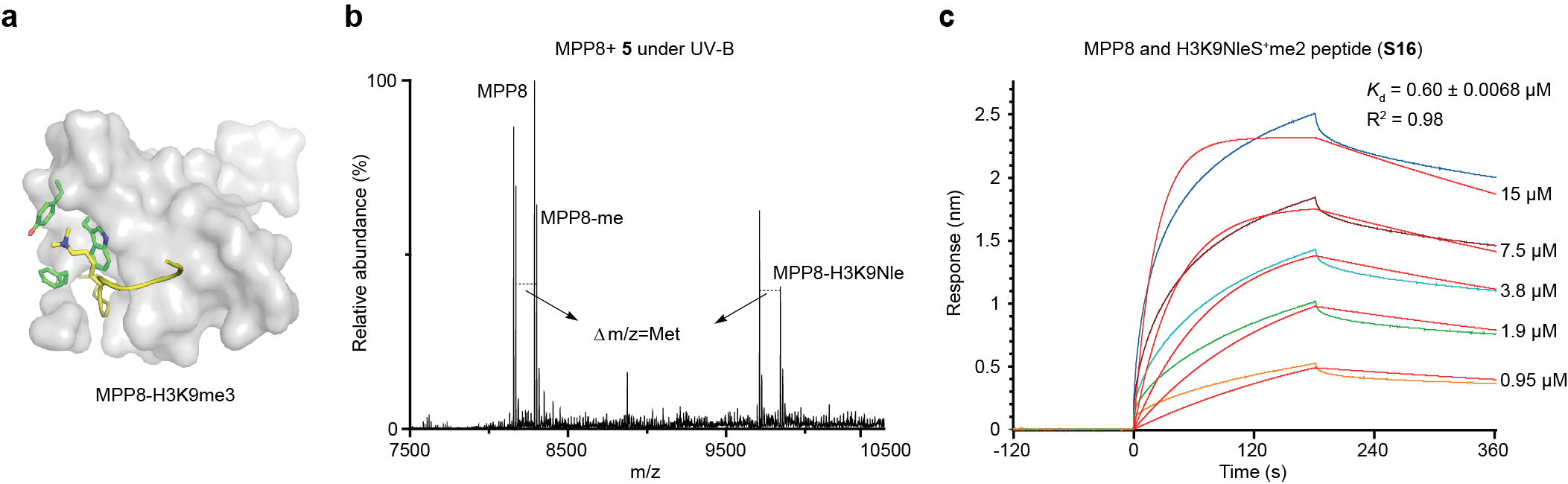
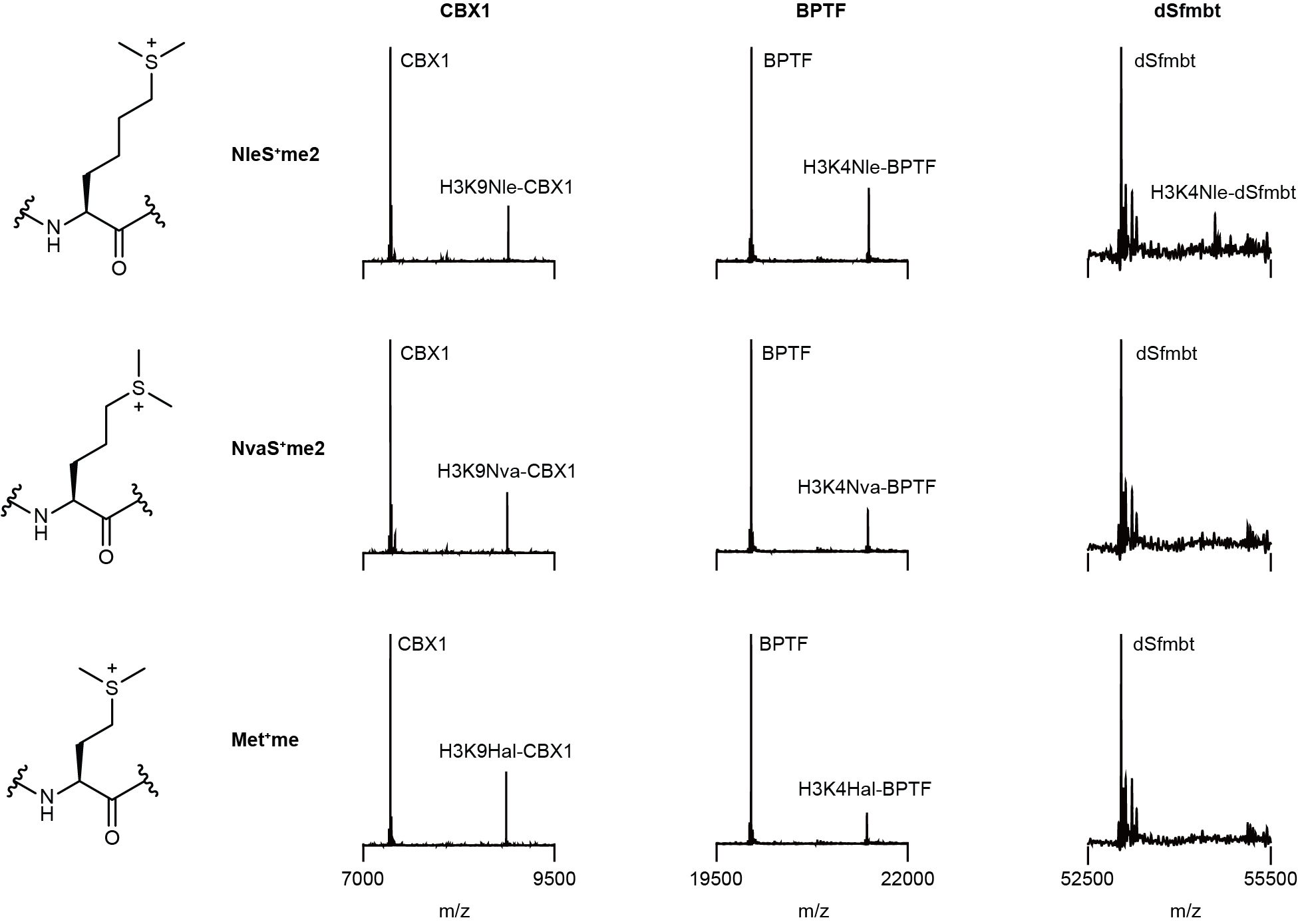
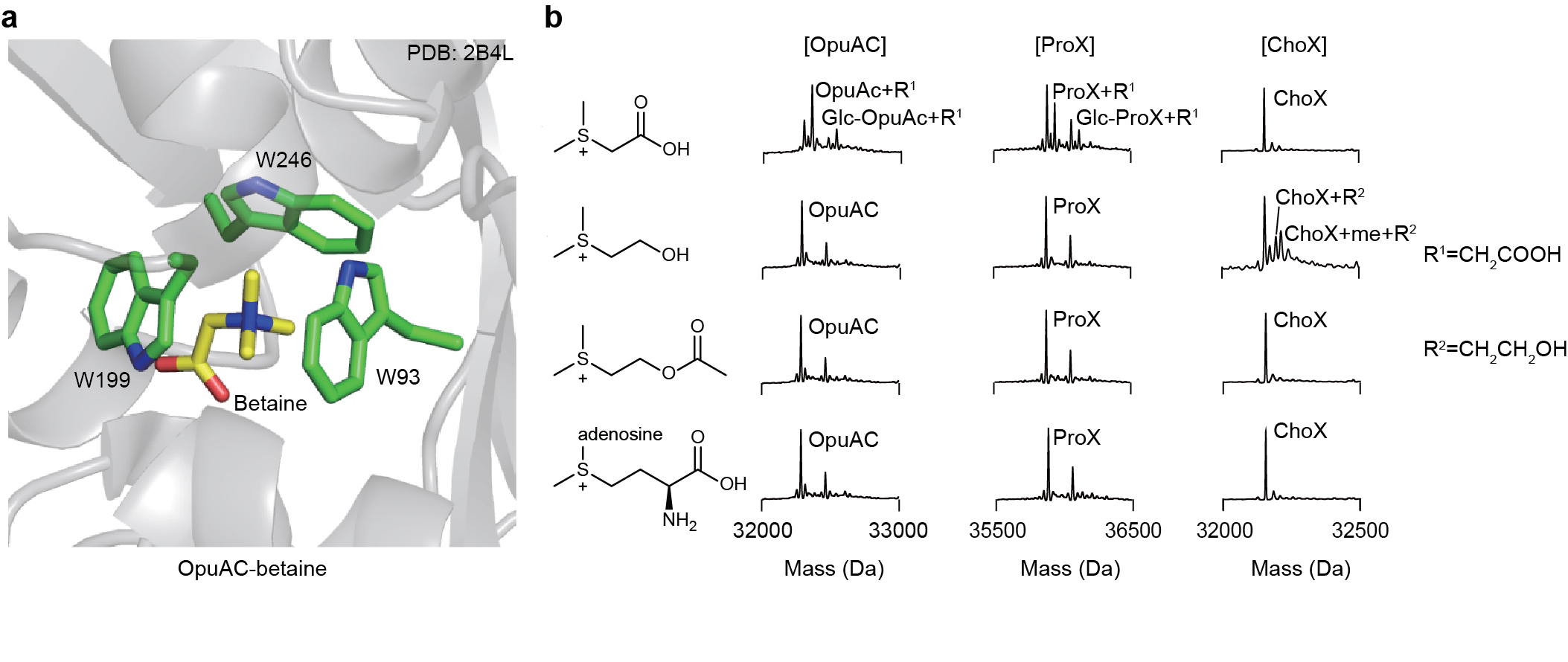
General Crosslinking Procedure. Reader protein in stock solution was diluted by HEPES buffer (100 mM HEPES (pH=7.5), 10 mM glutathione) and mixed with sulfonium peptides in ice bath for 15 min. The solution (total volume: 50 μL) was transferred into a 96-well plate and irradiated for 5 min in ice bath using a 302 nm UV lamp (under 305 nm long-pass filter). The reaction mixture was later analyzed by UPLC-Q-TOF-MS with a gradient of 5-95% B over 4 min and 95% B over 2 min. The analytic yield was calculated based on mass peak areas of starting material and products from deconvolution of the mass spectrometry data. Yield = Ap/As where Ap is the peak area of peptide-conjugated product and As is sum of all protein peak areas including the residual starting material, peptidyl product, methyl products, and other side product (if any).
Kinetics study of the crosslinking between H3K9NleS+me2 and CBX1.
10 μM CBX1 in HEPES buffer (100 mM HEPES, pH=7.5) was mixed with H3K9NleS+me2 peptide (5) of serial concentrations. All the samples were incubated on ice for 15 min and then transferred into a 96-well plate for 3 min photo crosslinking as described in General Crosslinking Procedure. According to integration of mass peaks from UPLC-Q-TOF-MS analysis, the initial reaction rate V0 was calculated from analytic yield of peptide-CBX1 conjugate. Finally, the processed data were fitted by the program GraphPad Prism (equation: Michaelis-Menten model). Next, CBX1 at different concentrations in HEPES buffer was mixed with 625 μM H3K9NleS+me2 peptide (5). After photo crosslinking at the same condition, the resulting mixtures were analyzed by UPLC-TOF. The data were processed by the program GraphPad Prism (equation: first order polynomial model (straight line)).
Investigation of nuclear methyllysine readers by sulfonium probes.
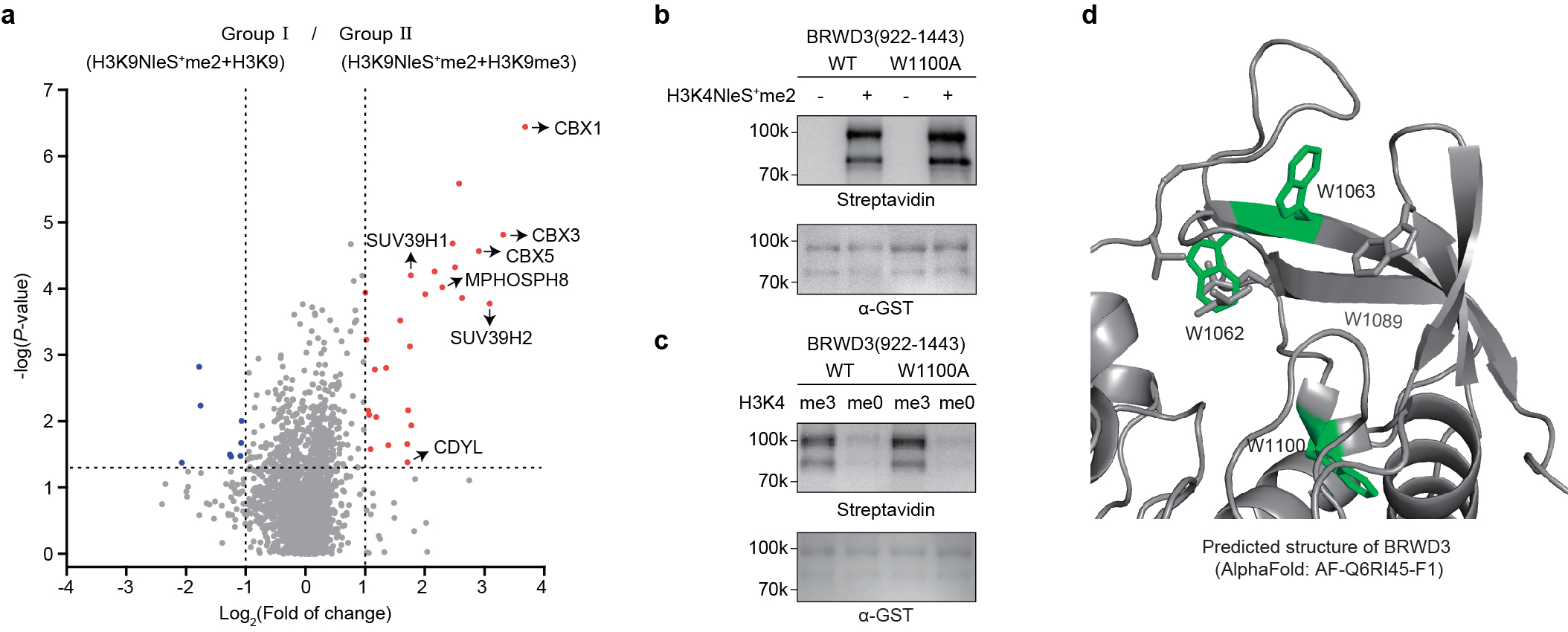
HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, C11965500CP) containing 10% fetal bovine serum (FBS), 10 U/m penicillin and 100 mg/mL streptomycin (HyClone, SV30010) at a humidified 37 ºC incubator with 5% CO2. After cells in ~90% confluency were harvested, the in situ crosslinking experiments were carried out referred to reported methods with some modifications58,59. Briefly, 5×106 Hela cells were lysed in hypotonic lysis buffer A (10 mM Tris, 15 mM NaCl, 1.5 mM MgCl2, 0.2 mM PMSF, pH 7.6) for 10 min on ice, then homogenized with 5 strokes of a loose pestle Dounce homogenizer. The nuclei were isolated by spinning 200 g for 5 min at 4 °C, and resuspended in crosslinking buffer (100 mM HEPES, 1.5 mM MgCl2, 150 mM KCl, 0.2 mM PMSF, pH 7.5). Next, sulfonium peptide probe (50 μM) and unmodified peptide (500 μM) were added into the isolated nuclei in group I. The same amount of sulfonium peptide and Kme3 peptide were added to group II nuclei. The mixture was incubated for 20 min followed by irradiation with UV-B light for 5 min on ice. After that, the excess peptides were removed via nuclei washing with crosslinking buffer twice. Next, the nuclei were resuspended with extraction buffer (25 mM HEPES, 1.5 mM MgCl2, 300 mM KCl, 1 mM EDTA, 0.1% NP40, 0.5 mM DTT, pH 7.5), were sheared by sonication with 8 cycles of a probe sonicator at 20% amplitude for 5 s on and 10 s off. After the removal of any insoluble materials by centrifugation, the nuclear proteins were loaded onto pierce streptavidin magnetic beads (Thermo Fisher, 88817), which were blocked and equilibrated with 1 mg/mL BSA and extraction buffer. The subsequent immunoprecipitation was carried out at 4 °C for 2 h with end-over-end rotation.
The beads were sequentially washed with extraction buffer twice, high salt buffer (25 mM HEPES, 1 M NaCl, pH 7.5), urea buffer (25 mM HEPES, 2 M urea, pH 7.5), TE buffer (25 mM HEPES, 1 mM EDTA, pH 7.5) and 50 mM ammonium bicarbonate at pH 8.0 twice. Next, trypsin Gold (2 μl of a 1 μg/μL stock) was added to the beads in 200 μl ammonium bicarbonate, and the on-beads digestion was performed overnight at 37 °C with 1500 rpm in a thermo mixer (IKA Matrix Orbital). Additional trypsin (1 μL of a 1 μg/μL stock) was added for another 2 h digestion. The supernatant was collected, and the beads were washed with ammonium bicarbonate. The combined supernatants were lyophilized.
The lyophilized samples were resuspended in 15 μL 0.1% formic acid for LC-MS/MS analysis. The peptides were separated by a 120 min gradient elution at a flow rate 0.300 µL/min with the Thermo Vanquish Neo integrated nano-HPLC system which is directly interfaced with the Thermo Exploris 480 mass spectrometer. The analytical column was a home-made fused silica capillary column (75 µm ID, 150 mm length; Upchurch, Oak Harbor, WA) packed with C-18 resin (300 A, 3 µm, Varian, Lexington, MA). Mobile phase A consisted of 0.1% formic acid in water, and mobile phase B consisted of 80% acetonitrile and 0.1% formic acid. The mass spectrometer was operated in the data-dependent acquisition mode using the Xcalibur 4.1 software and there is a single full-scan mass spectrum in the Orbitrap (350-1800 m/z, 60,000 resolution) followed by 20 data-dependent MS/MS scans at 30% normalized collision energy. The AGC target was set as 5e4, and the maximum injection time was 50 ms. Each mass spectrum was analyzed using the Thermo Xcalibur Qual Browser and Proteome Discoverer for the database searching against the Mus musculus proteome database downloaded from UniProtKB (UP000000589) containing 55,311 proteins and the Homo sapiens proteome database downloaded from UniProtKB (UP000005640) containing 80,581 proteins as of October 18, 2022, respectively. The Sequest search parameters included a 10 ppm precursor mass tolerance, 0.02 Da fragment ion tolerance, and up to 2 internal cleavage sites. Fixed modifications included cysteine alkylation, and the methionine oxidation was variable modification. Peptides were filtered with 1% false discovery rate (FDR). These values were subsequently adjusted for two-tail t-test. Protein ratios with a ratio greater than 2.0 and a P-value less than 0.05 were considered significant. The mass spectrometry data have been deposited in the ProteomeXchange Consortium repository as an open-source dataset under the identifier PXD051693.
Crosslinking mass spectrometry (XL-MS) of peptidyl conjugate by sulfonium probes.
XL-MS of recombinant readers protein. Crosslinking mixtures of BPTF with H3K4NleS+me2 peptide (S14), JMJD2A with H3K4NleS+me2 peptide (S14), JMJD2A with H4K20NleS+me2 peptide (S15), mORC1 with H4K20NleS+me2 peptide (S15), CBX1 with H3K9NleS+me2 peptide (S16) were reduced with 10.0 mM dithiothreitol at 37°C for 1 hour, followed by alkylation with 20 mM iodoacetamide in an aqueous solution for 30 minutes at room temperature in the dark. The beads were then washed three times with 50 mM ammonium bicarbonate at pH 8.0. Digestion was performed with Glu-C at an enzyme-to-protein ratio of 1:30 (w/w) overnight at 37°C. Afterward, the beads were washed twice with 50 mM ammonium bicarbonate. Subsequently, 10 μL of the sample was digested using a mixture of Lys-C and trypsin enzymes at enzyme-to-protein ratios of 1:50 (w/w) and 1:30 (w/w) respectively, at 37°C for 10 hours. Finally, the sample was desalted using homemade Venusil XBP C18 (5 μm, 150 Å) desalting tips prior to LC-MS/MS analysis.
The sampless were initially re-dissolved in a solution containing 0.1% formic acid (FA). These samples were analyzed using an Easy-nLC 1000 system coupled with a Q-Exactive mass spectrometer (Thermo Fisher Scientific, USA). The control of the mass spectrometer and data collection were managed using the Q-Exactive Tune Application (2.8 SP1 Build 2806) and Thermo Scientific Xcalibur software (v3.1.66.10), respectively. The samples were automatically loaded onto a C18 reversed-phase (RP) trap column (150 μm i.d. × 3 cm) and separated on a C18 capillary column (150 μm i.d. × 15 cm), which was in-house packed with ReproSil-Pur C18-AQ particles (1.9μm, 120 Å). The mobile phases used were buffer A (98% H2O, 2% ACN, 0.1% FA) and buffer B (2% H2O, 98% ACN, 0.1% FA). The separation gradient was programmed as follows: 2–10% B over 10 min, 10–23% B over 50 min, 23–40% B over 20 min, 40–80% B over 2 min, followed by a hold at 80% B for 13 min. The mass spectrometry settings included data-dependent acquisition, full MS resolution of 70,000 at m/z 200, a scan range of 300–1800, MS1 automatic gain control (AGC) target of 3e6, MS1 maximum injection time (IT) of 60 ms, MS/MS resolution of 17,500 at m/z 200, a fixed first mass of 110 m/z, MS/MS AGC target of 5e4, MS/MS maximum IT of 60 ms, a loop count of 20, an isolation window of 2.0 m/z, higher-energy collision dissociation (HCD) with a normalized collision energy (NCE) of 28, charge exclusion for unassigned, 1, and >8 charges, an intensity threshold of 1000, and a dynamic exclusion of 18 s.
XL-MS of nuclei samples. Swelled 3.5x106 HeLa cells with 12 mL 1xRSB buffer and incubated on ice for 15 min. Collected crude nucleus by 4 ºC, 200 g, 5min. Leave about 1.5 mL supernatant to resuspended pellet and homogenized with 5 strokes. Then, nucleuses were pelleted at 4 ºC, 200g for 5min, discarded supernatant and resuspended nucleuses with crosslinking buffer (100 mM HEPES, pH 7.5, 150 mM KCl, 1.5 mM MgCl2, added 0.2 mM PMSF before use). The nuclei were incubated with sulfonium probes (50 μM) and incubated on ice for 15 min followed with irradiation (302 nm UV light, 305 nm filter, ice, 5 min). The un-crosslinked probes were removed by twice crosslinking buffer washing (4 ºC, 200 g, 5 min). The nuclei were resuspended in extraction buffer (25 mM HEPES, 1.5 mM MgCl2, 300 mM KCl, 1 mM EDTA, 0.1% NP40, 0.5 mM DTT, pH 7.5), and nuclear proteins were extracted through sonication, using 8 cycles of a probe sonicator at 20% amplitude for 5 seconds on and 10 seconds off. Following centrifugation to remove any insoluble materials, the nuclear protein was loaded onto Pierce high-capacity streptavidin agarose resin (Thermo Fisher, 20359) and immunoprecipitated at room temperature for 3 hours with end-over-end rotation.
The beads were washed sequentially three times with extraction buffer and twice with 50 mM ammonium bicarbonate. Next, 10.0 mM dithiothreitol was added and incubated at 37°C for 1 hour, followed by the addition of a 20.0 mM iodoacetamide aqueous solution, which was incubated at room temperature for 30 minutes in the dark. After centrifugation, the beads were washed twice with 50 mM ammonium bicarbonate. Glu-C (30 μg) was then added to the beads in 600 μL ammonium bicarbonate, and on-beads digestion was performed overnight at 37°C. The beads were washed with 50 mM ammonium bicarbonate twice. Then additional Lys-C (15 μg) and trypsin Gold (30 μg) were added, and digestion continued at 37°C for another 10 hours. After digestion, the supernatant was collected, and the beads were washed twice with ammonium bicarbonate and twice with 20% acetonitrile in water, with the wash solutions also being collected. The combined supernatants were then lyophilized.
For peptide, homemade C18 tips (5 μm, 100 Å; Durashell) were employed for desalting and fractionation. After activating and equilibrating the C18 tips, the peptides were loaded onto the tips and washed three times with solvent A (H2O with ammonia added, pH 10.0) for desalting. The elution solvent, solvent B (acetonitrile with ammonia added, pH 10.0), was used to create 9 eluates (6%, 9%, 12%, 15%, 18%, 21%, 25%, 30%, 80% B), which were combined into fractions as follows: 6% and 25% for fraction 1, 9% and 30% for fraction 2, 12% and 80% for fraction 3, and 15%, 18%, and 21% for fractions 4, 5, and 6, respectively. Finally, the fractionated peptides were lyophilized and subsequently analyzed using nano-LC-MS/MS.
The samples were re-dissolved in a solution containing 0.1% formic acid (FA) and analyzed using an Easy-nano LC 1200 system, coupled to an Orbitrap Exploris 480 instrument equipped with a FAIMS Pro device (Thermo Fisher Scientific). During FAIMS separations, temperatures of the inner and outer electrodes were maintained at 100 °C, and the total carrier gas flow rate was set to 4.0 L/min. Compensation voltage (CV) values for each injection were -45 and -65. The mass spectrometry analysis utilized two mobile phases: mobile phase A (0.1% FA in HPLC-grade H2O) and mobile phase B (acetonitrile with 20% H2O and 0.1% FA). Peptides were separated on a C18 capillary column (150 μm i.d. × 150 mm) packed with C18 silica particles (1.9 μm, 120 Å) from Dr. Maisch GmbH, with the column heated to 55°C and a flow rate of 600 nL/min. The gradient started at 5% B, increasing to 9% B over 10 minutes, then from 9% to 20% B over the next 35 minutes, followed by an increase from 20% to 35% B over 40 minutes, and finally from 35% to 48% B over 25 minutes. The mass spectrometer operated in positive ion mode with data-dependent acquisition (DDA). MS1 scans were performed at a resolution of 60,000 (at m/z 200) from m/z 350 to 1500, and MS2 scans at a resolution of 15,000. The maximum injection times were set to 20 ms for MS1 and 30 ms for MS2. In each full MS scan, the most intense ions with charge states from 3 to 7 were selected for sequencing, using an isolation window of 1.6 m/z and a cycle time of 2 seconds. Fragmentation of precursor ions was achieved using HCD mode with a normalized collision energy of 30.
Mass data analysis. For the analysis of crosslinked peptides, the raw files were processed using the OpenUaa software60, with searches conducted against the protein FASTA file of model readers protein and quantitative proteomics-derived readers. Carbamidomethyl (C) was considered as fixed modification; Oxidation (M) and acetylation of the protein N-terminus were considered as variable modification. The maximum number of missed cleavage sites was set to three. The precursor mass tolerance and fragment mass tolerance were both set at 20 p.p.m. The search results were filtered using a false discovery rate (FDR) of 5% at the peptide-spectrum match (PSM) level. The mass spectrometry data have been deposited in the ProteomeXchange Consortium repository as an open-source dataset under the identifier PXD049149.
Characterization of BRWD3 as a H3K4me3 reader
Crosslinking between recombinant BRWD3 and H3K4NleS+me2 peptide. 3μM GST-tagged BRWD3 (922-1443), BRWD3 (922-1443_W1062A&W1063A), BRWD3 (922-1443_W1100A), or BRWD3 (1140-1443) was mixed with H3K4NleS+me2 peptide (S14) (100 μM) for 10 min crosslinking by General Crosslinking Procedure. The crosslinking mixtures were analyzed by western blot using pierce™ high sensitivity streptavidin-HRP, and total proteins were analyzed using α-GST antibody.
Analysis of BRWD3-H3K4me3 peptide interaction by chemical crosslinker. 4 μM putative reader protein with GST tag was mixed with 50 μM H3K4me3 or unmodified H3 peptide with biotin tag. The mixture was incubated on ice for 20 min before adding 1 mM chemical crosslinker (Bis-PEG5-NHS ester). The crosslinking reaction was allowed to proceed for 20 seconds at room temperature, followed by quenching using 100 mM Tris for another 15 min. The resulting sample was analyzed by western blot using HRP-conjugated Streptavidin and α-GST antibody respectively.
Crosslinking BRWD3 in nuclei by H3K4NleS+me2 peptide. Human BRWD3(922-1450) was cloned into pcDNA3.1 expression vector with an N-terminal 3xFLAG tag. Endofree plasmid was prepared by following manufacturer’s instruction (CWBIO, CW2107M). HeLa cells were seeded in 100 mm dishes with about 40% confluent one day before and were later transfected with the plasmid when growth to 70-80% confluent. 15 μg plasmid was diluted by 250 μL Opti-MEM medium and mixed with 30 μL P3000. Next, 12 μL lipo3000 in 250 μL medium was added and incubated for 15 min at room temperature. The about 500 μL plasmid-lipid complex solution was later added to 10 mL HeLa cell culture in 100 mm dish for BRWD overexpression. After 24h, the cells were harvested for following crosslinking reactions.
1x106 HeLa cells were swelled with 4 mL 1xRSB buffer (10 mM Tris-HCl, pH 8.0, 15 mM KCl, 1.5 mM MgCl2, fresh 0.2 mM PMSF) and incubated on ice for 15 min. Crude nuclei were collected by 200 g centrifugation at 4 ºC and resuspended with 1.5 mL buffer for homogenization with 2 strokes. The nuclei were next pelleted and resuspended with crosslinking buffer (100 mM HEPES, pH 7.5, 150 mM KCl, 1.5 mM MgCl2, fresh 0.2 mM PMSF). The nuclei were incubated with unmodified peptide (500 μM) or Kme3 peptide (500 μM) on ice for 8 min. Next, sulfonium probe S14 (50 μM) was added into each tube for 15 min incubation on ice. After 5 min photo crosslinking, the remaining peptides were removed by twice washing with crosslinking buffer. Next, the nuclei were resuspended with sonicate buffer (25 mM HEPES, pH 7.5, 1.5 mM MgCl2, 300 mM KCl, 1 mM EDTA, 0.1% NP40, fresh 0.5 mM DTT) for subsequent sonication (5 s on, 10 s off, 8 cycles, AMP: 25%). Centrifugation (4 ºC, 13000 g, 10 min). After centrifugation, the nuclear protein was loaded onto pierce streptavidin magnetic beads (Thermo Fisher, 88817), which were equilibrated and blocked with extraction buffer and 1 mg/mL BSA. The immunoprecipitant was carried out at 4 °C for 2 h with end-over-end rotation. The protein bound beads were washed with sonicate buffer, high salt buffer (50 mM Tris, 1M KCl, pH 7.5), urea buffer (50 mM Tris, pH 7.5, 2M urea), EDTA buffer (50 mM Tris, pH 7.5, 1 mM EDTA), Tris-HCl buffer (50 mM Tris, pH 7.5) and 1xPBS buffer for one time. Next, the beads were boiled with 40 μL 1xSDS buffer at 95 ºC for 30 min and the enriched proteins were analyzed by western blot.
Immunoprecipitation (IP). Streptavidin magnetic beads (500 μg) were preincubated with biotin-H3Kme0 peptide (1.2 nmol) or biotin-H3Kme3 peptide (1.2 nmol) at 4 °C for 2 h with end-over-end rotation. The peptide bound beads were washed with sonicate buffer, urea buffer and sonicate buffer twice. Next, nuclei were extracted from HeLa cells with BRWD overexpression and sheared by sonication by the same procedure above. The soluble nuclear proteins were equally separated into peptide bound beads. The immunoprecipitations were carried out at 4 °C for 2 h with end-over-end rotation. The protein bound beads were washed with sonicate buffer, high salt buffer, EDTA buffer, and 1xPBS buffer twice. Next, the beads were boiled with 40 μL 1xSDS buffer at 95 ºC for 30 min and enriched proteins were analyzed by western blot.
Methods-only references
58. Yang, Q., Gao, Y., Liu, X., Xiao, Y. & Wu, M. A general method to edit histone H3 modifications on chromatin via sortase-mediated metathesis. Angew. Chem. Int. Ed. 61, e202209945 (2022).
59. Burton, A. J., Haugbro, M., Gates, L. A., Bagert, J. D., Allis, C. D. & Muir, T. W. In situ chromatin interactomics using a chemical bait and trap approach. Nat. Chem. 12, 520-527 (2020).
60. Liu, C., Wu, T., Shu, X., Li, S. T., Wang, D. R., Wang, N. et al. Identification of protein direct interactome with genetic code expansion and search engine OpenUaa. Adv. Biol. 5, e2000308 (2021).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}