Chemistry
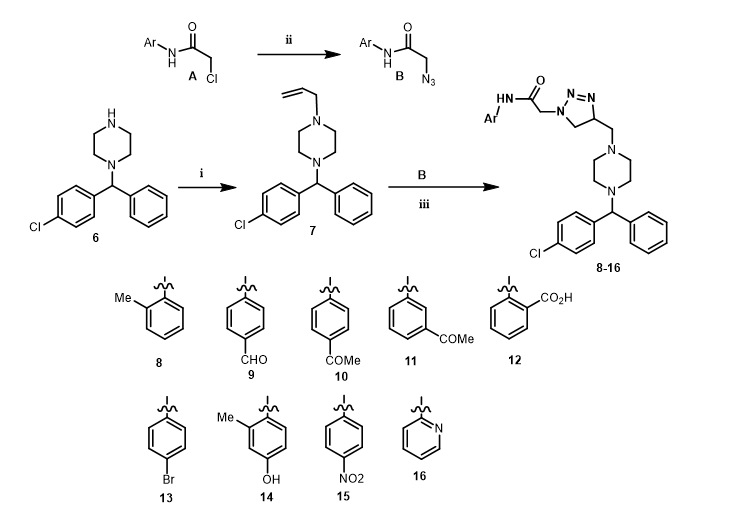
1-Ally-4-((4-chlorophenyl)(phenyl)methyl)piperazine (7), derived from the reaction of 1-benzhydryl piperazine (6) and allyl bromide in the presence K2CO3/KI, has been selected as a crucial intermediate for synthesising novel 1-benzhydryl piperazine-based Δ2-1,2,3-triazoline hybrids. A yield of 79% was obtained for the formation of 7. Diez-Gonzales et al. [32] have documented the synthesis of Δ2-1,2,3- triazoline derivatives using deep eutectic solvent (DES) via the cycloaddition of aryl azides with olefines. In this study, our object is to investigate the synthesis of novel benzhydrylpiperazine analogues, inspired by the work of Diez-Gonales and his group. Additionally, we aim to assess the antiproliferative activity against various cancer cells. Thus, one-pot cycloaddition of 7 with various substituted 2-zido-N-aryl-acetamides (B): 2-azido-N-(o-tolyl)acetamide, 2-azido-N-(4-formylphenyl)acetamide, N-(4-acetylphenyl)-2-azidoacetamide, N-(3-acetylphenyl)-2-azidoacetamide, 2-(2-azidoacetamido)benzoic acid, 2-azido-N-(4-cyanophenyl) acetamide, 2-azido-N-(4-bromophenyl)acetamide, 2-azido-N-(4-hydroxyphenyl) acetamide, 2-azido-N-(4-nitrophenyl)acetamide, 2-azido-N-(pyridine-2-yl) acetamide, prepared in situe from the chloro analogue (A), in the presence of DES (choline chloride/urea 1:2, 0.5 M) at 80 oC gave the benzhydrazylpiperazine Δ2-1,2,3-triazoline analogues 8–16 in 45–60% yield. (Scheme 1).
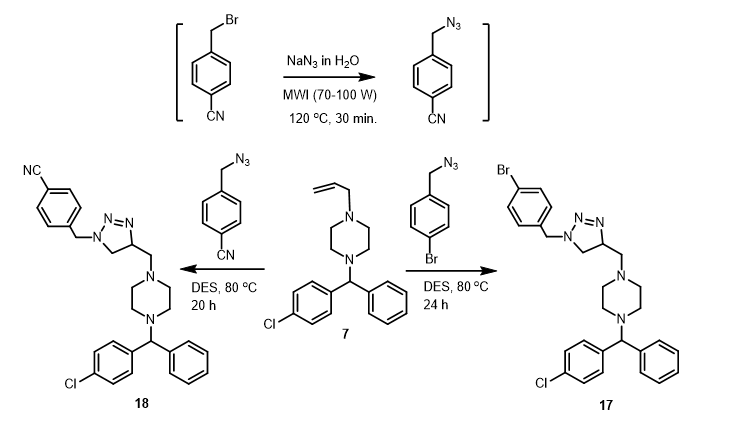
Similary, treatment of 7 with 4-bromobenzyl azide, which was prepared in situ from 4-(bromomethyl)benzonitrile, under the same conditions resulted in the formation of Δ2-1,2,3-triazoline derivatives 17 and 18 in 59 and 52 yield, respectively (Scheme 2).
The structures of 8–18 were confirmed by their IR, 1H, 13C and 2D NMR spectra. The Δ2-1,2,3-triazoline scaffold showed a silimar pattern. In the 1H NMR spectra of 8–18, the singlet in the range d = 2.40–2.50 ppm were assigned to CH2 protons, while multiplet in the range 2.68–2.95 and 2.73-3.00 ppm were attributed to the eight piperazine protons. Htriazoline-4’ appeared as broad singlet in the range d = 3.69–4.04 ppm, while Htriazoline-4’ resonated as multiplet or broad singlet in the range d = 2.59–2.73 ppm. Hbridge proton appeared as singlet in the range d = 5.12–5.22 ppm. The singlet in the range d = 3.41–3.57 ppm assigned to CH2O protons of 8–16, whereas the two singlets at d = 6.57 ppm attributed to CH2Ph-Br and CH2Ph-CN of 17 and 18. The other aliphatic and aromatic protons were fully analyzed (c.f. Experimental section). In the 13C NMR spectra of 8–16, the carbonyl carbon atoms resonated in the ranges d = 165.9-172.7 ppm, while the resonances at d = 195.4, 197.8 and 176.6 ppm assigned to carbonyl carbon atoms of 2xCOMe and CO2H, respectively. The resonances in the ranges d = 44.5–51.7 and 51.1–53.0 ppm were attributed to piperazine carbon atoms, while the signals in the range d = 52.6–56.5 ppm were assigned to CH2 carbon atoms. Carbon-4’ of Δ2-1,2,3-triazoline ring
appeared in the range d = 60.3–61.2 ppm, whereas carbon-5’ of the same moiety resonated in the range d = 62.2–68.9 ppm. The resonances in the range d = 73.0-74.6 ppm were attributed to the bridge carbon atoms. CH2O carbon atom of 8–16 appeared in the range d = 56.5–59.2 ppm. The other aliphatic and aromatic carbon atoms were fully analyzed (c.f. Experimental section).
Compound 10 was selected for more detailed NMR studies. In the gradient-selected HMBC spectrum [33] of 10, the carbonyl carbon atom at δ 172.5 ppm showed a 2JC,H correlation with methylene protons (CH2O) at δΗ 3.41 ppm, while the methylene protons (CH2) adjacent to piperazine ring at δΗ 2.53 ppm showed a 3JC,H correlation with carbon-5’ of Δ2-1,2,3-triazoline ring at δC 63.5 ppm. In addition, a 2JC,H coupling between Htriazoline-4’ at δH 3.82 ppm and the CH2 carbon atom adjacent to piperazine ring at δC 52.9 ppm was observed. Furthermore, methylene proton at carbon-5’ of Δ2-1,2,3-triazoline scaffold at δH 2.69 ppm showed a 2JC,H coupling with carbon-4’ of Δ2-1,2,3-triazoline ring at δC 61.0 ppm (Fig. 2).
Biological evaluations
In vitro cytotoxic activity
The anticancer activity of the newly synthesized compounds 8–18 as evaluated on a diverse range of of human cancer cell lines including pancreatic adenocarcinoma (Capan)-1, colorectal carcinoma (HCT-116), glioblastoma (LN-229), acute lymphoblastic leukemia (DND-41), acute myeloid leukemia (HL-60), chronic myeloid leukemia (K-562), and non-Hodgkin lymphoma (Z-138) cancer cells (Table 1). The screening was conducted using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) viability assay [34] with etoposide and nocodazole used as reference drugs for comparison. Compound 11 exhibited the highest activity among the derivatives, inhibiting the proliferation of the selected tested human cancer cell lines HL-60, Z138, and DND-41 with IC50 values of 16.80, 18.50 and 19.20 µM, respectively, while it demonstrated a higher micromolar IC50 values towards the Capan-1, HTC-116, LN229 and K562 cell lines with IC50 values of 40.90, 36.25, 43.90 and 36.10 µM, respectively. Compound 10 displayed IC50 values of 19.90, 18.00 and 18.50 µM against the cell lines HL-60, Z138 and DND-41. It exhibited moderate activity against Capan-1, HTC-116, LN229 and K562 cell lines (ranging from 36.10 to 43.90 mM). In addition, compound 13 showed IC50 value of 19.90 µM against DND-41 cell line, while compound 14 showed activity against HL-60 cell line with IC50 value of 27.75 µM. Furthermore, compound 16 demonstrated selectivity towards the HL-60 and Z-138 cell lines with IC50 values of 27.10 and 22.95 µM, respectively.
The SAR study indicated that the anticancer activity of compounds 7–18 depwnded on the substituents of the aryl amide ring. Compound 11, which featured an acetyl substituent at position meta of the phenyl-amide ring, displayed the most significant antitumoral activity among the tested compounds, especially against the HL-60 cancer cell line. Moreover, compound 11 displayed marginally lower antitumoral activity against DND-41 and Z138 cell lines compared to the HL-60 cell line. These finding provide encouragement to synthesize various substituted 1,2,3-triazoline derivatives, as they may enhance the antiproliferative effects on different cancer cell lines.
Mode of action
Acute myeloid leukemia (AML) is a malignant disease of the hematopoietic system characterized by uncontrolled cell proliferation, impaired cell differentiation, and infiltration of bone marrow, peripheral blood, or other tissues [35]. A crucial protein involved in various cellular processes, such as cell proliferation, apoptosis, and transcription, is Akt, also known as protein kinase B (PKB) or Rac. In AML, Akt1, a specific serine/threonine-specific protein kinase, plays a significant role by activating the PI3K/AKT pathways and serving as a key signaling protein [36]. As a result, Akt1 is commonly targeted in AML therapy, and multiple Akt1 inhibitors have been developed for this purpose. The mode of action of compound 11 can be attributed to its ability to inhibit the PI3K/AKT pathway, as assured by predictive docking studies.
Table 1
In vitro cytotoxicitya of some benzhydrylpiperqazine analogues given as IC50b in µM
| Comp. | | | | IC50 (µM) | | | | |
| | | | | Cell Lines | | | | |
| | Capan-1 | HCT-116 | LN229 | DND-41 | HL-60 | K562 | Z138 | |
| 8 | 44.30 | 51.25 | 58.45 | 38.85 | 38.45 | 44.15 | 36.85 | |
| 9 | 46.00 | 38.90 | 52.05 | 30.15 | 31.30 | 37.75 | 28.40 | |
| 10 | 46.35 | 37.45 | 35.40 | 18.50 | 19.90 | 39.40 | 18.00 | |
| 11 | 40.90 | 36.25 | 43.90 | 19.20 | 16.80 | 36.10 | 18.50 | |
| 12 | 45.15 | 45.95 | 44.45 | 39.45 | 42.10 | 44.65 | 41.45 | |
| 13 | 41.95 | 40.00 | 37.70 | 19.90 | 43.6 | 40.55 | 40.65 | |
| 14 | 45.25 | 29.00 | 34.05 | 33.9 | 27.75 | 41.95 | 37.90 | |
| 15 | 47.40 | 51.25 | 41.55 | 36.70 | 38.75 | 46.00 | 39.90 | |
| 16 | 45.25 | 42.75 | 40.70 | 31.70 | 27.10 | 36.9 | 22.95 | |
| 17 | 44.35 | 51.25 | 46.95 | 37.10 | 42.55 | 49.75 | 39.90 | |
| 18 | 45.00 | 43.70 | 47.45 | 53.60 | 50.0 | 47.3 | 32.9 | |
| ETP | 0.10 | 3.50 | 1.65 | 2.00 | 0.30 | 2.15 | 0.35 | |
| NDZ | 0.02 | 0.135 | 0.085 | 0.025 | 0.025 | 0.03 | 0.025 | |
ETP: Etoposide, NDZ: Nocodazole. a Cytotoxicity as IC50 for each cell line is the concentration of tested compound with reduced by 50% the optical density of treated cells with respect to untreated cells using the MTT assay. b Data represent mean values of at least two independent experiments.
Antioxidant activity
The evaluation of antioxidant activity was carried using DPPH (1,1-diphenyl-2-picryl hydrazyl) radical scavenging assay [37], with ascorbic acid serving as a positive control for comparison. This method is based on the reduction of DPPH radicals solution in the presence of hydrogen donating antioxidant, resulting in the formation of the non-radical form DPPH-H. The results of antioxidant activity of compounds 8–18 are presented in Table 2. The percentage inhibition of radical scavenging was measured at various concentrations (1000, 900, 800, 700, 600, 500, 250, 125, 62.5 and 31.25 µM), representing the concentration required to scavenge 50% of DPPH radicals. As shown in Table 2, most compounds exhibited low or negligible antioxidant activity in the DPPH assay, with inhibition percentages ranging from 0 to 33.7 µM at a solution concentration of 31.25 µM. Compound 13 exhibited the highest antioxidant activity, inhibiting 33.7% at a concentration of 31.25 and 73.2 µM at a concentration of 1000 µM, compared to the standard ascorbic acid (93.6 and 99.7%, respectively). Compounds 17 and 18, containing 4-bromobenzyl and 4-cyanobenzyl-1,2,3-triazoline groups, exhibited antioxidant activity of 25.4 and 30.9% at a concentration of 31.25 µM, respectively, and inhibition of 86.2 and 84.8% at a concentration of 1000 µM 54.4 and 54.5%, respectively. Specifically, among the series, only compound 13 displayed moderate DPPH scavenging activity. Therefore, 13 can be considered as a lead compound for further development in terms of DPPH scavenging activity.
Table 2
Antioxidant activity of new benzhydrylpiperazine analogues 8–18
| Compd. % Inhibition conc. |
| | 1000 | 900 | 800 | 700 | 600 | 500 | 250 | 125 | 62.5 | 31.25 |
| 8 | 84.5 | 81.8 | 80.9 | 77.9 | 72.4 | 50.3 | 46.1 | 39.2 | 33.7 | 21.8 |
| 9 | 8.8 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 10 | 68.8 | 58.6 | 34.0 | 25.4 | 1.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 11 | 39.2 | 22.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 12 | 17.1 | 1.7 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| 13 | 73.2 | 71.0 | 69.9 | 68.2 | 62.7 | 57.5 | 54.1 | 49.4 | 47.2 | 33.7 |
| 14 | 76.0 | 75.7 | 72.7 | 68.5 | 63.8 | 58.3 | 53.9 | 45.3 | 32.3 | 20.7 |
| 15 | 44.8 | 32.9 | 24.6 | 17.4 | 13.3 | 6.1 | 0.0 | 0.0 | 0.0 | 0.0 |
| 16 | 85.1 | 77.6 | 75.4 | 69.1 | 64.1 | 58.6 | 56.6 | 44.8 | 30.9 | 8.8 |
| 17 | 86.2 | 79.3 | 74.9 | 72.1 | 69.1 | 61.3 | 55.8 | 47.0 | 29.3 | 25.4 |
| 18 | 84.8 | 79.8 | 75.4 | 72.7 | 69.1 | 64.1 | 58.6 | 47.8 | 45.3 | 30.9 |
| A.A. | 99.7 | 99.2 | 98.3 | 98.1 | 97.5 | 97.2 | 96.1 | 95.3 | 94.8 | 93.6 |
Docking study analysis
The objective of conducting molecular docking calculations is to anticipate the most propable binding between specific protein and a ligand. In this study, Autodock4 [38] was employed for the docking calculations, and the obtained docking results were visualized and analyzed using MGL Tools. The receptor molecule was prepared by elimination water molecules from their structures and then subjected to correction and 3D protonation refinment.
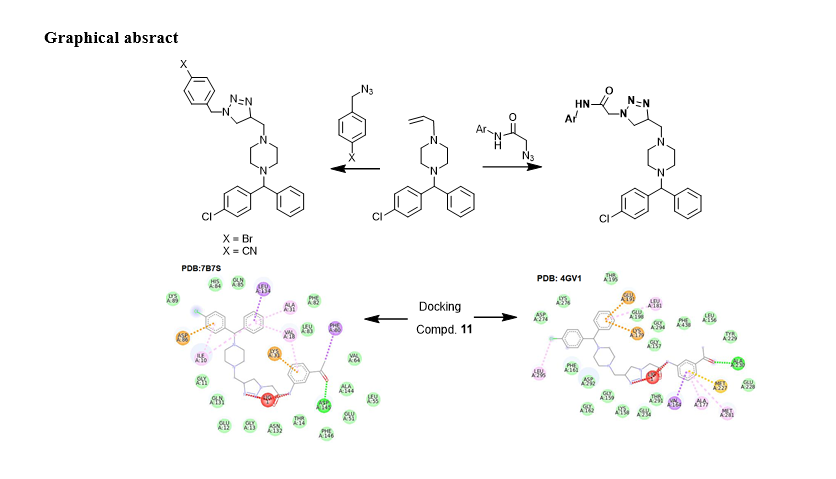
To explore a potential anticancer lead derivative, compound 11 was chosen for molecular docking investigations. The three-dimensional structures of acute myeloid leukemia (cell cycle-related protein CDK2/cyclin A2, PDB: 7B7S), and protein kinase Akt1 PKB alpha, PDB: 4GV1) were used for the docking procedure using the Autodock4 program. The ligand of interest was ranked based on the highest energy of the best conformations. The calculated binding energy scores for compound 11 were − 10.00 kcal mol-1 for both 4GV1 and 7B7S. These results suggest selective binding of this analogue to the active site of the protein receptor pockets in 7B7S, and 4GV1.
In Fig. 3 (A), the docking results depicted the appropriate positioning of compound 11 for its binding with the protein receptors of cell cycle-related protein CDK2/cyclin A2 (PDB: 7B7S). Two hydrogen bonds were formed between the N-3 and N-4 of the tetrazole moiety and Cys694. Furthermore, a π-π stacking interaction occurred between the phenyl ring of the benzyl group and the phenyl residue of Arg834. Additionally, a π-H interaction was observed between the phenyl group of the tetrazole ring and Leu616. Surrounding compound 11, various non-bonded amino acid residues, including Asp829, Asn816, Arg815, Glus831, Cys828, Val624, Gly697, Lys644, and 644, were observed in the receptor structure.
In Fig. 3 (B), the docking analysis revealed that compound 11 binds to the protein kinase Akt1 PKB alpha (PDB: 4GV1). A hydrogen bond (1.876 Å) formed between the oxygen of the carbonyl group attached to the phenyl residue and Ala230. Additionally, the phenyl group of the benzhydryl piperazine scaffold displayed π - cation and π - anion interactions with Glu191 and Lys179, along with a π - alkyl interaction with Leu181. Furthermore, the phenyl group, which was conjugated with
a 1,2,3-triazoline moiety, exhibited a π - σ interaction with Val164 and π - alkyl interactions with both Met281 and Ala177. The same phenyl group also showed a π - sulfur interaction with Met227.
The chloro residue demonstrated an alkyl interaction with Leu295. In the vicinity of compound 11, there were non-bonded amino acids present in the receptor structure, including Asp274, Lys276, Thr195, Gly157, Glys294, Tyr229, Thr291, Gly159, Asp292, and Phe161. The docked complex exhibited acceptable RMSD scores.
Experimental section
General information
Melting points were determined using a Mel-Temp device melting point apparatus and are uncorrected. The IR spectra were recorded, on an FT-IR Spectrophotometer (Thermo Nicolet Corp. USA), using KBr discs. 1H and 13C NMR spectra were acquired on Bruker Avance (400 MHz) (1H) and 75 MHz (13C) spectrometers, using DMSOd6 solvent containing tetramethylsilane as an internal standard (chemical shifts expressed in δ in ppm). The progress of reactions were monitored by thin layer chromatography (eluent: hexane-EtOAc 4: 1), and the spots were visualized using iodine and U.V. Microwave supported reactions were performed in microwave reaction vials (2.5 mL) with Teflon septum and an aluminium crimp top, utilizing a microwave cavity operating at 2.4 GHz and temperature ranging from 0-300 oC .
1-Allyl-4-((4-chlorophenyl)(phenyl)methyl)piperazine (7)
To a solution of 1-benzhydryl piperazine (6) (820 mg, 2.86 mmol) in dry acetone (15 mL) was added allyl bromide (0.30 mL, mmol) and anhyd. K2CO3 (789 mg, 5.72 mmol) followed by the addition of KI (0.28 mg, 0.17 mmol) and the reaction mixture was heated under reflux 6 h. After the completion of the reaction (followed by TLC), the solvent was removed under reduced pressure; the residue was poured into ice cold water and extracted with CHCl3 (3x20 mL). The combined organic layer was dried (MgSO4), filtered and evaporated to dryness. The crude product was purified on short column of SiO2 (5 g) using hexane-EtOAc (3:2) as eluent. To give 7 (738 mg, 79%) as yellow solid product, m.p.: 83–85 oC, Rf =0.34. FT-IR (KBr, cm− 1): 2964, 2809 3059, 3025, 3100 (C-H), 1646 (C = Callyl), 1599, 1486 (C = Carom.). 1H NMR (400 MHz, DMSO) δ 2.71, 2.99 (m, 8H, Hpiperazine), 3.12 (s, 2H, CH2-CH = CH2), 5.14 (s, 1H, Hbridge), 5.16, 5.20 (d, J = 12.0 Hz, 2H, CH2-CH = CH2), 5.70 (dd, J = 5.0, 12.1 Hz, 1H, H-15), 7.17–7.77 (m, 9H, Harom.). 13C NMR (100 MHz, DMSO) δ 44.6, 52.9 (Cpiperazine), 61.0 (CH2-CH = CH2), 74.1 (Cbridge), 118.3 (CH2CH = CH2), 127.5, 128.0, 129.0, 129.1, 129.8, 131.8, 134.4, 141.6, 142.7 (Carom.+CH2-CH = CH2). Anal. calc. for C20H23ClN2 (326.87): C, 73.49; H, 7.09; N, 8.57. Found: C, 73.41; H, 7.01; N, 8.49
General procedure for the synthesis of 1,2,3-triazoline derivatives (8–15)
A mixture of 2-chloro-N-aryl-acetamide or 4-substituted benzyl bromide derivatives (1.0 mmol) and sodium azide (84.5 mg, 1.3 mmol) dissolved in water (5 mL) was subjected to microwave irradiation (70–100 W) at 120 oC for a duration of 30 min. After cooling, the mixture was extracted with CHCl3 (3x15 mL), and the combined organic extracts was dried (MgSO4), filtered and evaporated to dryness. The collected azide derivative was used directly for the next step. To a solution of 7 (287 mg, 0.92 mmol) in deep eutectic solvent (choline chloride / urea 1:2, 0.5 M) was added the appropriate aryl azide (1.84 mmol) and the rection mixture was stirred at 80 oC for 20–24 h. After colling to room temperature, water (10 mL) was added and extracted with EtOAc (3x10 mL). The combined organic extracted was dried (MgSO4), filtered an evaporated to dryness. The crude product was purified by SiO2 column (20 g) using, in gradient, hexane (0–40%) and EtOAc as eluent to give the desired product.
2-(4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(o-tolyl)acetamide (8)
Yield: 290 mg (56%) as a dark brown semisolid. Rf =0.63. FT-IR (KBr, cm− 1): 3390 (N-H), 3082, 3058, 3027, 2973, 2936, 2854 (C-H), 1645 (C = O), 1520, 1486 (C = C). 1HNMR (400 MHz, DMSO) δ 2.52 (CH2), 2.59 (br s., 2H, Htriazoline-5’), 2.95, 2.73 (m, 8H, Hpiperazine), 3.52 (s, 2H, CH2CO), 3.99 (br s., 1H, Htriazoline-4’), 5.22 (s, 1H, Hbridge), 7.18–7.58 (m, 13H, Harom.), 9.91 (br., 1H, NH). 13C NMR (100 MHz, DMSO) δ 9.0 (Me), 45.8, 51.1 (Cpiperazine), 52.6 (CH2), 57.9 (CH2-CO), 60.5 (CHtriazoline-4’), 62.2 (CH2pyrazoline-5’), 73.0 (Cbridge), 125.1, 125.5, 127.5, 129.8, 131.8, 132.2, 133.5, 134.23, 141.9, 142.3 (Carom.), 172.7 (C = O). Anal. calc. for C29H33ClN6O (517.07): C, 67.36; H, 6.43; N, 16.25. Found: C, 67.18; H, 6.27; N, 16.08
2-(4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(4-formylphenyl)acetamide (9)
Yield: 260 mg (49), as a brown solid, m.p.: 63–65 oC, Rf =0.68. FT-IR (KBr, cm− 1): 3059, 2973, 2935, 2854 (C-H), 1721, 1640 (C = O), 1596, 1523, 1486 (C = C). 1H NMR (400 MHz, DMSO) δ 2.53 (CH2), 2.62 (m, 2H, Htriazoline-5’), 2.71, 2.89 (m, 8H, Hpiperazine), 3.57 (s, 2H, CH2CO), 3.75 (br s., 1H, Htriazoline-4’), 5.20 (s, 1H, Hbridge), 7.18–7.58 (m, 13H, Harom.), 9.58 (s, 1H, NH), 9.89 (s, 1H, CHO). 13C NMR (100 MHz, DMSO) δ 46.0, 52.5 (Cpiperazine), 53.5 (CH2), 59.2 (CH2-CO), 60.3 (CHtriazoline-4’), 62.4 (CH2pyrazoline-5’), 74.2 (Cbridge), 125.4, 127.6, 128.0, 129.0, 129.1, 129.8, 131.9, 132.3, 131.9, 133.5, 141.4, 142.1, 142.5 (Carom.), 172.5 (C = O), 190.0 (CHO). Anal. calc. for C29H31ClN6O2 (531.06): C, 65.59; H, 6.68; N, 15.83. Found: C, 65.39; H, 6.47; N, 15.62.
N-(4-Acetylphenyl)-2-(4-((4-((4-chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)acetamide (10)
Yield: 327 mg, (60%) as a brown semisolid, Rf =0.74. FT-IR (KBr, cm− 1): 3396 (NH), 3057, 2974, 2936, 2854 (C-H), 1731, 1651 (C = O), 1597, 1556, 1486 (C = C). 1H NMR (400 MHz, DMSO) δ 2.50 (s, 3H, COMe), 2.53 (s, 2H, CH2), 2.69 (m, 2H, Htriazoline-5’), 2.71, 2.98 (m, 8H, Hpiperazine), 3.41 (s, 2H, CH2CO), 3.82 ((br s., 1H, Htriazoline-4’), 5.15 (s, 1H, Hbridge),7.21–7.67 (m, 13H, Harom.), 10.73 (br s., 1H, NH). 13C NMR (100 MHz, DMSO) δ 26.3 (COMe), 45.8, 51.6 (Cpiperazine), 52.9 (CH2), 57.4 (CH2-CO), 61.0 (CHtriazoline-4’), 63.5 (CHtriazoline-5’), 74.5 (Cbridge), 121.6, 127.5, 129.0, 129.3, 129.8, 131.0, 135.3, 141.9, 142.4, 142.8 (Carom.) 172.5, 195.4 (2xC = O). Anal. calc. for C30H33ClN6O2 (545.08): C, 66.11; H, 6.10; N, 15.42. Found: C, 66.00; H, 5.99; N, 15.32
N-(3-Acetylphenyl)-2-(4-((4-((4-chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)acetamide (11)
Yield: 311 mg (57%) as a semisolid, Rf = 0.74. FT-IR (KBr, cm− 1): 3375 (NH), 3058, 2929, 2821 (C-H), 1720, 1678 (C = O), 1600 (C = C). 1H NMR (400 MHz, DMSO) δ 2.51 (s, 3H, COMe), 2.56 (s, 2H, CH2), 2.68 (m, 2H, Htriazoline-5’), 2.71, 2.95 (m, 8H, Hpiperazine), 3.43 (s, 2H, CH2CO), 3.89 ((br s., 1H, Htriazoline-4’), 5.13 ((s., 1H, Hbridge), 7.09–7.67 (m, 13H, Harom.), 10.53 (br., 1H, NH). 13C NMR (100 MHz, DMSO) δ 27.2 (COMe), 45.9, 51.4 (Cpiperazine), 52.8 (CH2), 57.4 (CH2-CO), 60.8 (CHtriazoline-4’), 68.9 (CHtriazoline-5’), 73.0 (Cbridge), 119.0, 125.4, 127.5,127.9, 129.0, 129.1, 129.3, 129.8, 131.8, 132.2, 138.1, 141.9, 142.3, 142.7 (Carom.), 176.2, 197.8 (2xC = O). Anal. calc. for C30H33ClN6O2 (545.08): C, 66.11; H, 6.10; N, 15.42. Found: C, 65.98; H, 5.99; N, 15.31
2-(2-(4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)acetamido)benzoic acid (12)
Yield: 246 mg (45%), as a brown amorphous, Rf = 0.86. FT-IR (KBr, cm− 1): 3059, 3028, 2974, 2937, 2824 (C-H), 1700, 1648 (C = O), 1612, 1582 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.40 (s, 2H, CH2), 2.68 (br s., 2H, Htriazolin-5’), 2.70, 2.98 (m, 8H, Hpiperazine), 3.42 (s, 2H, CH2CO), 3.82 (br s., 1H, Htriazolin-4’), 5.14 (s., 1H, Hbridge), 7.20, 7.22, 7.24, 7.29, 7.31, 7.33, 7.35, 7.38, 7.40, 7.42, 7.45, 7.46, 7.48 (m, 13H, Harom.), 10.61 (s, 1H, NH), 13.36 (s,1H, OH). 13C NMR (100 MHz, DMSO) δ 45.8, 51.7 (Cpiperazine), 53.0 (CH2), 57.4 (CH2-CO), 61.1 (CHtriazoline-4’), 65.7. (CHtriazoline-5’), 74.6 (Cbridge), 115.0 (Carom.-1’), 118.1 (Carom.-3’), 125.4, 127.5, 128.0, 129.0, 129.1, 129.8, 131.7, 131.8, 135.7, 141.5, 142.4, 142.8 (Carom.), 170.3 (C = O), 176.6 (CO2H). Anal. calc. for C29H31ClN6O3 (547.06): C, 63.67; H, 5.71; N, 15.36. Found: C, 63.60; H, 5.64; N, 15.30
N-(4-bromophenyl)-2-(4-((4-((4-chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)acetamide (13)
Yield: 291 mg (50%), as a brown amorphous, Rf = 0.58. FT-IR (KBr, cm− 1) : 3374 (N-H), 3083, 3059, 3027, 2927, 2853, 2811 (C-H), 1646 (C = O), 1600, 1487 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.42 (CH2), 2.60 (br s., 2H, Htriazolin-5’), 2.71, 2.95 (m, 8H, Hpiperazine), 3.43 (s, 2H, CH2CO), 3.69 ((br s., 1H, Htriazoline-4’), 5.12 (s., 1H, Hbridge), 7.16–7.77 (m, 13H, Harom.), 10.32 (br s., 1H, NH). 13C NMR (100 MHz, DMSO) δ 45.8, 51.6 (Cpiperazine), 53.0 (CH2), 57.4 (CH2-CO), 61.1 (CHtriazoline-4’), 66.9 (CHtriazoline-5’), 74.5 (Cbridge), 118.3, 121.6, 127.5, 127.9, 128.0, 129.0, 129.3, 129.4, 129.8, 131.8, 132.3, 135.4, 141.9, 142.4 (Carom.), 172.5 (C = O). Anal. calc. for C28H30BrClN6O (581.94): C, 57.79; H, 5.20; N, 14.44. Found: C, 57.67; H, 5.08; N, 14.21
2-(4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(4-hydroxyphenyl)acetamide (14)
Yield: 265 mg (51%), as a yellow-brown solid, m.p.: 99–101 oC, Rf = 0.91. FT-IR (KBr, cm− 1): 3400 (OH), 3204 (NH), 3061, 2964, 2926, 2814 (C-H), 1676 (C = O), 1606 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.52 (CH2), 2.64 (br s., 2H, Htriazolin-5’), 2.73, 2.98 (m, 8H, Hpiperazine), 3.49 (s, 2H, CH2CO), 3.72 ((br s., 1H, Htriazoline-4’), 5.16 (s., 1H, Hbridge), 6.74–7.53 (m, 13H, Harom.), 9.47 (s, 1H, NH), 10.02 (s, 1H, OH). 13C NMR (100 MHz, DMSO) δ 48.7, 51.8 (Cpiperazine), 53.1 (CH2), 56.5 (CH2-CO), 61.2 (CHtriazoline-4’), 67.8 (CHtriazoline-5’), 74.6 (Cbridge), 115.6, 121.6, 127.5, 128.0, 129.0, 129.1, 129.8, 130.5, 131.8, 142.8, 142.4 (Carom.), 154.2 (C-OH), 165.9 (C = O). Anal. calc. for C28H31ClN6O2 (519.05): C, 64.79; H, 6.02; N, 16.19. Found: C, 64.70; H, 5.93; N, 16.11
2-(4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(4-nitrophenyl)acetamide (15)
Yield: 323 mg (59%), as a brown solid, m.p.: 91.94 oC, Rf = 0.77. FT-IR (KBr, cm− 1): 3059, 3028, 2924, 2814 (C-H), 1634 (C = O), 1597, 1556 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.48 (CH2), 2.73 (br s., 2H, Htriazolin-5’), 2.68, 2.93 (m, 8H, Hpiperazine), 3.43 (s, 2H, CH2CO), 3.84 ((br s., 1H, Htriazoline-4’), 5.14 ((s., 1H, Hbridge), 6.74–8.26 (m, 13H, Harom.), 10.52 (br s., 1H, NH). 13C NMR (100 MHz, DMSO) δ 51.7, 53.0 (Cpiperazine), 56.5 (CH2), 57.4 (CH2-CO), 61.1 (CHtriazoline-4’), 63.0 (CHtriazoline-5’), 74.6 (Cbridge), 118.2, 125.4, 127.5, 129.0, 129.1, 129.3, 129.8, 132.2, 141.9, 142.4, 142.8 (Carom.), 172.6 (C = O). Anal. calc. for C28H30ClN7O3 (548.04): C, 61.37; H, 5.52; N, 17.89. Found: C, 61.18; H, 5.35; N, 17.70.
2-(4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)-N-(pyridin-2-yl)acetamide (16)
Yield: 242 mg (48%), as a light green amorphous, Rf = 0.63. FT-IR (KBr, cm− 1): 3391 (NH), 3060, 3026, 2960, 2928, 2808 (C-H), 1645 (C = O), 1599 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.45 (CH2), 2.70 (br s., 2H, Htriazolin-5’), 2.72, 3.00 (m, 8H, Hpiperazine), 3.43 (s, 2H, CH2CO), 4.04 ((br s., 1H, Htriazoline-4’), 5.20 ((s., 1H, Hbridge), 7.13–7.77 (m, 12H, Harom.), 8.22 (m, 1H, Hpyridine-6’). 13C NMR (100 MHz, DMSO) δ 44.6, 51.5 (Cpiperazine), 52.9 (CH2), 57.4 (CH2-CO), 60.9 (CHtriazoline-4’), 63.0 (CHtriazoline-5’), 74.5 (Cbridge), 118.5 (Cpyridin-3’), 125.5, 127.5, 128.0, 129.0, 129.8, 131.8, 138.1, 141.9, 142.4, 146.3 (Carom.+Cpyridin.), 146.3 (Cpyridine-6’), 158.5 (Cpyridin.-1’), 171.3 (C = O). Anal. calc. for C27H30ClN7O (504.04): C, 64.34; H, 6.00; N, 19.45 Found: C, 64.19; H, 5.87; N, 19.30
1-((1-(4-Bromobenzyl)-4,5-dihydro-1H-1,2,3-triazol-4-yl)methyl)-4-((4-chlorophenyl) (phenyl)methyl)piperazine (17)
Yield: 318 mg (59%), as a brown amorphous, Rf = 0.47. FT-IR (KBr, cm− 1): 3050, 3020, 2960, 2933, 2812 (C-H), 1596 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.52 (CH2), 2.71 (br s., 2H, Htriazolin-5’), 2.78, 2.91 (m, 8H, Hpiperazine), 3.75 ((br s., 1H, Htriazoline-4’), 5.16 (s, 1H, Hbridge), 6.57 (s, 2H, CH2-Ph-Br), 7.16–7.72 (m, 13H, Harom.). 13C NMR (100 MHz, DMSO) δ 44.5, 51.8 (Cpiperazine), 56.5 (CH2 + CH2-Ph-Br), 61.1 (Ctriazoline-4’), 63.8 (Ctriazoline-5’), 74.6 (Cbridge), 120.3, 127.5, 128.0, 131.7, 132.1, 135.9, 142.4, 142.8 (Carom.). Anal. calc. for C27H29BrClN5 (538.92): C, 60.18; H, 5.42; N, 13.00. Found: C, 60.02; H, 5.26; N, 12.83
4-((4-((4-((4-Chlorophenyl)(phenyl)methyl)piperazin-1-yl)methyl)-4,5-dihydro-1H-1,2,3-triazol-1-yl)methyl)benzonitrile (18)
Yield: 252 mg (52%), as a brown amorphous, Rf = 0.50. FT-IR (KBr, cm− 1): 3095, 2931, 2813 (C-H), 2228 (CN), 1617, 1510, 1472 (C = C). 1H-NMR (400 MHz, DMSO) δ 2.50 (CH2), 2.69 (br s., 2H, Htriazolin-5’), 2.72, 2.92 (m, 8H, Hpiperazine), 3.85 ((br s., 1H, Htriazoline-4’), 5.17 (s., 1H, Hbridge), 6.57 (s, 2H, CH2-Ph-Br), 7.25–7.85 (m, 13H, Harom.). 13C NMR (100 MHz, DMSO) δ 44.6, 51.8 (Cpiperazine), 53.1 (CH2 + CH2-Ph-Br), 61.1 (CHtriazoline-4’), 64.6 (CHtriazoline-5’), 74.6 (Cbridge), 111.2 (C-CN), 117.9 (CN), 127.5, 128.0, 129.0, 129.5, 129.8, 131.7, 133.1, 135.9, 141.9, 142.5 (Carom.). Anal. calc. for C28H29ClN6 (485.03): C, 69.34; H, 6.03; N, 17.33. Found: C, 69.24; H, 5.93; N, 17.22.
Biological assays
Anticancer activity in vitro
Cancer Cell Lines
The human cancer cell lines, including Capan-1, HCT-116, LN-229, NCI-H460, HL-60, K-562, H and Z-138 cancer cells, were sourced from the American Type Culture Collection (ATCC, Manassas, VA, USA) for this manuscript, Additionally, the DND-41 cell line was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ Leibniz-Institut, Germany). The culture media, except for those used for other cell lines which were purchased from Sigma, were acquired from Gibco Life Technologies, USA, and supplemented with 10% fetal bovine serum (HyClone, GE Healthcare Life Sciences, USA),
Proliferation Assays
For adherent cell lines (LN-229, HCT-116, NCI-H460, and Capan-1 cells), 384-well tissue culture plates (Greiner) were seeded at a density ranging from 500 to 1500 cells per well. Following overnight incubation, the cells were treated with seven different concentrations of the test compounds, ranging from 100 to 0.006 µM. On the other hand, suspension cell lines (HL-60, K-562, Z-138, and DND-41) were seeded in 384-well culture plates with the test compounds at the same concentration points, and cell densities ranging from 2500 to 5500 cells per well. The cells were then incubated with the compounds for 72 h. and subsequently analyzed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) reagent (Promega), following the manufacturer’s instructions. Absorbance measurments were measured at 490 nm using a SpectraMax Plus 384 (Molecular Devices), and the OD values were utilized to determine the 50% inhibitory concentration (IC50). The compounds underwent testing in two independent experiments. For HeLa cells, seeding was performed in 96-well microtiter plates with a cell density raging from 1 × 104 to 3 × 104 cells/mL, depending on the doubling times specific to the cell line. Test agents were added at five 10-fold dilutions (10 − 8 to 10 − 4 M), with working dilutions freshly prepared on the day of testing. After 72 h. of incubation, the cell growth rate was assessed using the MTS assay as previously described [34]. Absorbance measurments was taken at 570 nm, and the OD values were used to calculate the 50% inhibitory concentration (IC50). Each test was performed in duplicate, across at least two individual experiments.
Antioxidant assay by using DPPH radical scavenging method.
The antioxidant activity of the synthesized compounds 3–10 was assessed using DPPH (1,1-diphenyl-2-picrylhydrazyl) free radical scavenging assay. Initially, a 0.2 mM solution of DPPH in EtOH at various concentrations (1000, 900, 800, 700, 600, 500, 250, 125, 62.5, 31.25 µg/mL). This mixture was vigorously shaken and left at room temperature for 30 min. Absorbance then measured at 517 nm using a UV-VIS spectrophotometer (UV-VIS Shimadzu). To determine the antioxidant activity of the tested compounds, the percentage of DPPH neutralization by them was calculated and compared to the standard antioxidant ascorbic acid. The percent DPPH scavenging effect (%) or % inhibition was calculated using following equation:
DPPH scavenging effect (%) or % inhibition = Ac(0) – AA(t)1 / Ac(0) × 100.
where Ac(0) represents the absorbance of the control at t = 0, and AA(t) is the absorbance of the antioxidant at t = 30 min. All measurements were performed in triplicate
Docking and virtual screening
Preparations of ligands and proteins.
The 3D structures of ligand 11 were first generated using Avogadro software (v. 1.0.1) [39] and saved in PDB format. Subsequently, the ligand was prepared by selecting torsions, and the structures were converted from PDB format to PDBQT format. This conversion was done using the MGLTools software, which also performed united atom Kollman charges, fragmental volumes, and solvation parameter calculations for both the proteins and the ligand. To ensure an accurate docking process, the ligand's structure was subjected to energy minimization using the MMFF94 force field. The native ligands and crystallographic water molecules were then removed from the PDB structures, and polar hydrogens were added before commencing with the docking.
Molecular docking simulations
The Autodock program was utilized for molecular docking simulations. Prior to docking, the protein structures were prepared using UCSF Chimera 1.15 [40]. For pose sampling in Autodock, the Lamarkian Genetic Algorithm (LGA) was employed, and the number of energy simulations was set to 2,500,000. The default scoring function was utilized to calculate the docking scores. To generate the necessary maps, Autogrid was employed. The resulting docking outcomes were visualized using Biovia Discovery Studio 2020 software [41], followed by an analysis of the docking results. To validate the method, all docking simulations were performed using the ligands found in the crystal structures, and they were successful in reproducing the ligand-protein interaction geometries. Figure 3 illustrates the image of the native ligand for 7B7S and 4GV1 in comparison to the redocked native ligand with AutoDock. The root-mean-square deviation of atomic positions (RMSD) for these cases was 0.400 and 0.4008 Å, respectively.
{kind=link}
{kind=link}
{kind=link}