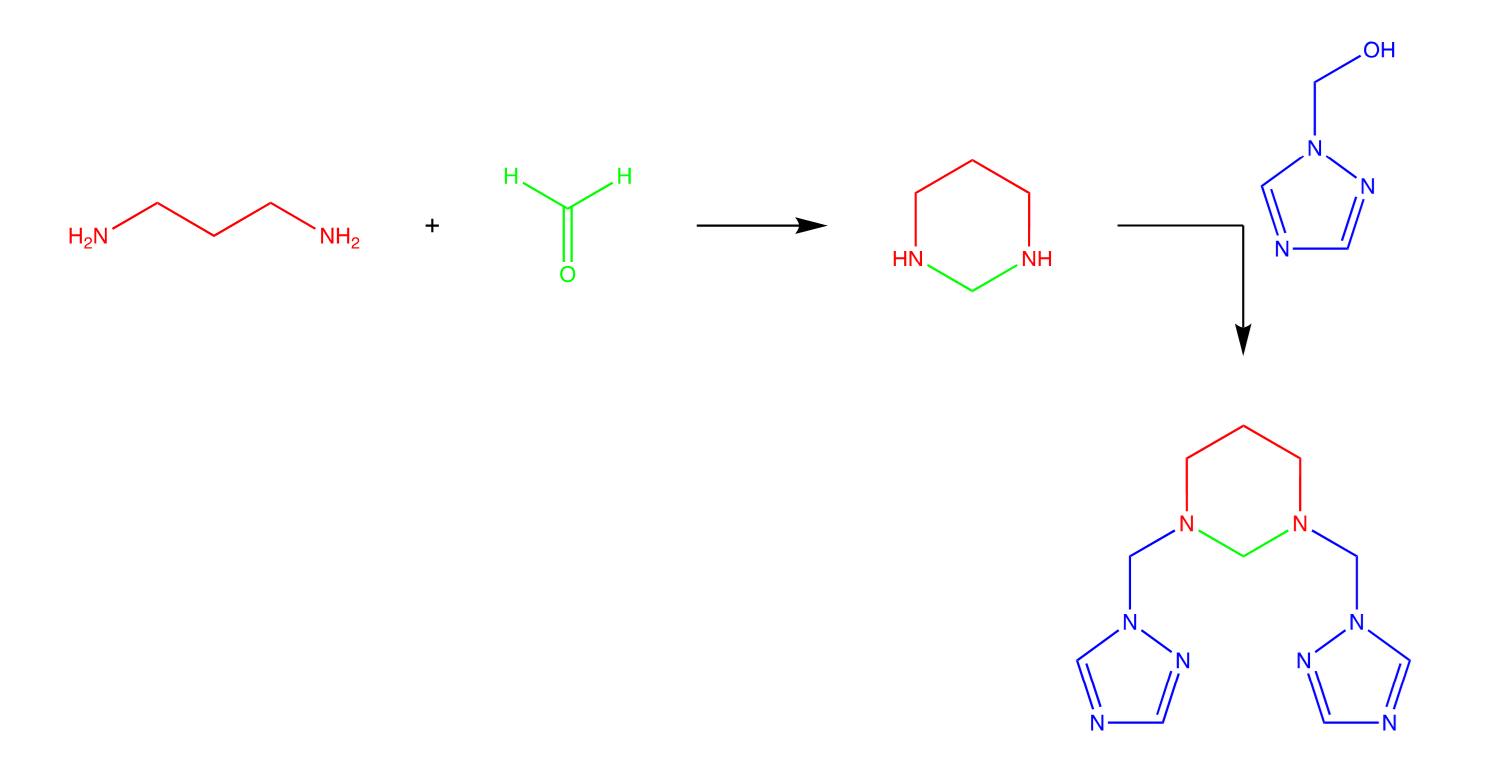
3.1. Synthesis
The creation of the methylene bridge by condensation of triazole and propane-1,3-diamine using an active carbonyl group of formaldehyde at a molar ratio of 2:1:4, leading to the formation of hetero-aliphatic rings. 1H-1,2,4-triazole in ethanol and excess formaldehyde solution (36.5%) was refluxed for 1 h, and stirring was continued at room temperature for 12 h. After the elimination of the solvent, the obtained residue of (1H-1,2,4-triazol-1-yl)methanol and formaldehyde was dissolved in acetonitrile with propane-1,3-diamine and refluxed for 1 h, 1,3-bis((1H-1,2,4-triazol-1-yl)methyl)hexahydropyrimidine was synthesized by mixing a tri-molecular combination of 1,3-propanediamine, formaldehyde, and (1H-1,2,4-triazol-1-yl)methanol in acetonitrile as solvent, under neutral conditions typically involves a one-pot reaction as shown in Scheme 1. This reaction can be a simple model for synthesizing various dihydropyrimidine derivatives.
One of the possible mechanisms of 1,3-bis((1H-1,2,4-triazol-1-yl)methyl)hexahydropyrimidine formation can be established as follows (Scheme 2); the 1,3-propanediamine contains two primary amine groups (-NH2) on each end of the molecule, acting as a nucleophile, with formaldehyde (H2CO).
It would appear that the reaction proceeds through a condensation step where one of the amine groups of 1,3-propanediamine attacks the carbonyl carbon of formaldehyde. This results in the formation of an imine intermediate. This intermediate can undergo intramolecular cyclization; the second nitrogen atom in the amine group attacks the imine's carbon, leading to the formation of a six-membered ring.
As a final step, both secondary amino groups can undergo substitution to introduce various substituents (R groups) by reacting with appropriate reagents such as (1H-1,2,4-triazol-1-yl)methanol, which would give the observed product. This step allows for synthesizing N, N’-substituted hexahydropyrimidine [25–27].
Therefore, the faster reaction rate observed between 1,3-propanediamine and formaldehyde can be attributed to the higher electrophilicity due to the electron-deficient carbonyl group of formaldehyde as compared to the lower electrophilicity and steric hindrance of the electrophilic center of (1H-1,2,4-triazol-1-yl)methanol.
This mechanism can be justified by the fact that we used an excess of (1H-1,2,4-triazol-1-yl)methanol. Nevertheless, the final product shows a monocondensation of the last reactant on each of both amines of the hexahydropyrimidine.
This result opens the way to new and numerous perspectives allowing access to new organic compounds.
3.2 Crystalline structure of L1
The L1 compound crystallizes in the Pnma space group with half a molecule in the asymmetric unit (a mirror passes through the atoms C4-C1). The molecular structure and crystalline packing are given in Fig. 4. No disorder was found at 150K and room temperature in the crystalline structure.
a) Molecular conformation
The molecule is somewhat flexible, with four torsional angles defining the relative orientation of the triazole groups; because of the symmetry of the experimental conformation, only two are reported in Table 3. The two triazole groups are more or less face-to-face (Fig. 4).
In the crystalline structural database, the first crystalline structure of a 1,3-derivative hexahydropyrimidine was found: 1,3-dicyanomethylhexahydropyrimidine (code CSD JULBUN [28] and its crystalline polymorph JULBUN01 [29]). The sole torsional angle comparison possible would be the equivalent of C3 N2 C5 N6, which appears to be -58.60° and 66.07° in JULBUN (and − 65.35° and 57.49° in JULBUN01) and ± 68.70(6)° in L1 at 150K (Table 2), leading to a relatively similar conformation (the -C ≡ N groups being almost parallel in JULBUN whereas the triazole groups are also nearly face to face in L1). Four additional crystalline structures were found in this database, the substituents in positions 1 and 3 on the hexahydropyrimidine ring being 4-bromophenol, phenol, 2-tert-butyl-4-methoxyphenol, 2,4-tert-butylphenol with CSD codes GAQYOO [30], LAQTEE [31], SAGXIJ [32], and XAYRIA [33], respectively. They exhibit, respectively the following torsional angles: (-69.76°, + 170.08°), (-62.63°, + 165.33°), ± 76.78° and ± 71.89. Consequently, GAQYOO and LAQTEE exhibit a similar conformation, but this conformation is different from the one of SAGXIJ and XAYRIA. At last, the conformation of SAGXIJ and XAYRIA is similar to L1 in the sense that the two phenol rings are on the same side of the hexahydropyrimidine ring, but they are not exactly face-to-face as in L1. This is reflected by the second torsional angle equivalent of C10 N6 C5 N2, which is ± 84.44(7)° in L1 at 150K (Table 2), ± 43.06° in SAGXIJ and ± 45.54° in XAYRIA.
The geometry of the isolated L1 molecule has been fully optimized with GAUSSIAN09 software and B3LYP/6-31G(d) method. The input model was obtained from the experimental crystallographic structure of L1. The optimized conformation of L1 remains relatively similar to the conformation in the crystalline structure despite the relative flexibility of the molecule; the internal symmetry is preserved (the mirror passing through C4-C1); see Table 4. The total energy is -828.660855310 Ha.
Several conformations were explored with GAUSSIAN09 by changing the torsion angles C3 N2 C5 N6 and C10 N6 C5 N2 (see Fig. 4a and Table 3) by 180°: the geometry was then fully optimized with the B3LYP/6-31G(d) method. Because of the molecule’s inherent symmetry, only five additional conformations can be found, see Fig. 5 and Table 5. Conformation 2 appears to have lower energy. Consequently, the observed conformation (conformation 1) is probably stabilized by the intermolecular interactions in the crystalline structure.
Table 3
Torsion angles (in °) characterizing the conformation of L1 at 150K and room temperature compared to the optimized torsion angles of conformation 1 (with B3LYP/6-31G(d) method).
| Temperature | 150 K | RT | optimized |
|---|
| C3 N2 C5 N6 | 68.70(6) | 68.54(10) | 63.88 |
| C10 N6 C5 N2 | -84.44(7) | -84.30(13) | -95.55 |
Table 4
Intermolecular close contacts in the L1 crystalline structure at 150K.
| D-H‧‧‧A | D-H (Å) | H‧‧‧A (Å) | D‧‧‧A (Å) | DHA (°) |
|---|
| C3 H3A N7 | 0.981(11) | 2.665(11) | 3.5049(7) | 143.9(8) |
| C5 H5B N9 | 0.973(10) | 2.686(10) | 3.6106(8) | 158.9(8) |
| C10 H10 N9 | 0.929(12) | 2.487(12) | 3.3577(8) | 156.3(10) |
| C8 H8 N2 | 0.964(11) | 2.639(11) | 3.5323(7) | 154.1(8) |
Table 5
Energies in Hartrees of different conformations of the L1 molecule.
| conformation | Energies in Ha | Relative energies (kJ.mol− 1) |
|---|
| 1 | -828.660853631 | 0.000 |
| 2 | -828.661419021 | -1.484 |
| 3 | -828.660135490 | + 1.885 |
| 4 | -828.658518828 | + 6.130 |
| 5 | -828.658577455 | + 5.976 |
| 6 | -828.659518836 | + 3.505 |
b) Crystal Packing and intermolecular interactions
Each molecule is involved in 8 weak intermolecular hydrogen bond networks C-H‧‧‧N with three different molecules (see Fig. 4b): the molecule being donor for four hydrogen bonds (involving the CH2 of the hexahydropyrimidine and the CH of the triazole groups) and acceptors for four hydrogen bonds (involving the nitrogen atoms of the hexahydropyrimidine and the two triazole groups). Because of the molecule’s symmetry, we must define only four kinds of hydrogen bonds (see Table 4 and Fig. 4b).
The C8H8‧‧‧N2 hydrogen bonds form an infinite chain along c (corresponding to the C11 (6) graph set [34–36]); because of the symmetry of the molecule, the same hydrogen bonds form a cyclic dimer (graph set R22(16)). Between these infinite chains of hydrogen bonds are the C10H10‧‧‧N9, forming infinite chains of hydrogen bonds along b (with graph set C11(11)). The C3H3A‧‧‧N7 and the C5H5B‧‧‧N9 hydrogen bonds form an infinite chain along a (corresponding to the C11 (6) graph set). Consequently, the four types of hydrogen bonds form a 3D network linking all the molecules (Fig. 4b).
c) Influence of the temperature
Cell parameters of L1 were followed by single crystal X-ray diffraction from 150K to ambient temperature (Table 2). No phase transition was observed. The volume (in Å3) of L1 as a function of temperature T (in K) can be fitted to a straight line, with r2 = 0.98:
V(T) = 0.1714 × T + 1207.6
It demonstrates that the volume increases linearly with temperature in the studied temperature range. It corresponds to an expansivity αv of 1.42 × 10− 4 K− 1 (Table 5), somewhat smaller than the average value 2 ×10− 4 K− 1 for small organic molecules [37], and the expansivity of another ligand poly(3,5-dimethylpyrazol-1-ylmethyl)benzene which also comprises C-H‧‧‧N hydrogen bonds (1.91 × 10− 4 K− 1) [38] but significantly larger than the one of another ligand with C-H‧‧‧N hydrogen bonds: N, N, N’, N’-tetrakis-[(1H,2,4-triazol-1-yl)methyl)]-ethane-1,2-diamine named TTYED (1.16× 10− 4 K− 1) [39].
The eigenvalues and eigenvectors of the thermal expansion tensor have been compiled in Table 6, while a graphical representation of the tensor at ambient temperature is presented in Fig. 6. Because the cell parameters increase linearly with temperature, the isobaric thermal expansion tensor is constant with temperature.
Table 6
Isobaric thermal tensor coefficients (in MK− 1) and orientation of the tensor in the unit cell.
| eigenvalues | eigenvectors |
|---|
| aV | 142(6) | a | b | c |
| a11 | 38(2) | 0.00 | 0.00 | 1.00 |
| a22 | 41(2) | -1.00 | 0.00 | 0.00 |
| a33 | 59(2) | 0.00 | -1.00 | 0.00 |
The aspherism coefficient has a value of 0.09, which is very close to zero, at least significantly small than the one of poly(3,5-dimethylpyrazol-1-ylmethyl)benzene (0.31) [38], ascorbic acid (0.47) [40], tienoxolol (0.50) [41] or the molecule N, N, N’, N’-tetrakis-[(1H,2,4-triazol-1-yl)methyl)]-ethane-1,2-diamine (0.58) [39], which indicates that the thermal expansion is more isotropic. And unlike these compounds, the expansivity of L1 is positive in all directions.
The maximum expansion occurs along e3 (or the b direction in an orthorhombic crystal), which is between 1.4 to 1.6 times larger than the expansion along the two other directions. This relatively high value indicates that e3 is the “soft direction” in the crystal. The minimum expansion is along e1 (or c direction); this can be referred to as the “hard direction” in the crystal. These two directions are parallel to a different infinite chain of hydrogen bonds. It can be concluded that the C8H8‧‧‧N2 hydrogen bonds parallel to c are weaker than the other hydrogen bonds in the crystalline structure. In contrast, the C10H10‧‧‧N9 hydrogen bonds parallel to b are the strongest. This is consistent with the experimental geometry of these hydrogen bonds (Table 4): C10H10‧‧‧N9 has the smallest A‧‧‧H distance and the hydrogen bond angle closest to the ideal 180° value.
Differential scanning calorimetry (DSC) was used to complete the thermal behavior of L1. To avoid the degradation of L1 that occurs at around 523 K (250°C), DSC studies were carried out below 473 K (200°C). The melting point of L1 was found to be 385.1 K (111.9°C) with an enthalpy of fusion of 132.6 J.g− 1 (32.9 kJ.mol− 1). Cooling the molten sample to 203 K (-70°C) with 5°C.min− 1 and reheating resulted in a glass transition at 258.1 K (-15.0°C; midpoint) on heating (Fig. 7). Subsequently, a large exothermic peak was observed related to recrystallization, which occurred between 336.3 K and around 368 K. After recrystallization, a melting peak was observed at 371.7 K (98.5°C, Fig. 7). The enthalpy associated with this melting transition was estimated at 39.8 J.g− 1 (9.9 kJ.mol− 1). This enthalpy value is probably underestimated because L1 has difficulty recrystallizing and because recrystallization and melting are concomitant. To our knowledge, there are no calorimetric data published on 1,3-disubstituted hexahydropyrimidine. However, analysis of melting point data from the literature [42] shows that the value obtained during the first heating is consistent with the range of values obtained for different N, N’-disubstituted hexahydropyrimidines between 60 and 204°C (average melting point = 130°C, median melting point = 121°C).
{kind=link}
{kind=link}