Plant material
Plant material used in this study is Cavendish Banana ( Musa spp. Cavendish; AAA Group cv. ‘Baxi’ ), the ‘Baxi’ banana is one banana cultivar that has been grown in China for many years and is one of the main banana cultivar in China.We obtained male flower buds of ‘Baxi’ banana from Institute of Fruit Tree Research, Guangdong Academy of Agricultural Sciences, Guangzhou, P. R. of China. ECS was induced by our laboratory in Institute of Fruit Tree Research, Guangdong Academy of Agricultural Sciences.
Banana protoplast preparation
Small and evenly distributed subculture of ECS for about 10 days was selected, the M2 medium was removed, and 10ml of enzymatic hydrolysate (3.0 % cellulose R-10, 1 % segregation enzyme R-10, 0.2 % pectinase Y-23, 15.2g/L KCl, 7.8g/L CaCl2, 100 mg/L MES, 10 % mannitol, pH 5.7) was added, and the cell were incubated in a shaking table at 50 rpm/min for 6-8 hours. The yield of protoplasts was observed by microscope. If the enzymatic hydrolysis was sufficient, the hydrolysate was diluted with 10 ml W5 solution and shaken for 10 s to separate the protoplasts. A 75 micron membrane was used to filter the enzymatic solution into a round bottom centrifugal tube. Centrifugation was performed at 100 g for 3min, and the supernatant was removed by pipette. Protoplasts were suspended in 15 ml W5 Solution, incubated on ice for 30 min and discard the supernatant.
gRNA design and vector construction
A total of 9 targets aiming at PDS gene in banana were designed by SnapGene software. The OsU3p vector was digested by Bsa1 and the fragments was recovered from agarose gel. The serial joint primers(Supplemental data set7) were linked to the recycled OsU3p vector by T4 ligase to generate OsU3p- PDSt1 to OsU3p- PDSt9, and then transformed into E. coli DH5α competent cells. After overnight culture at 37 ℃, single colonies verified by sequencing were inoculated in the LB liquid medium with ampicillin. After overnight culture at 37 ℃ 220 rpm, plasmids were extracted.
CRISPR/Cas12a vector construction
A total of 11 targets aiming at PDS gene in banana were designed by SnapGene software. The Cas12a vector was digested by Bsa1 and the fragments was recovered from agarose gel. The serial joint primers(Supplemental data set8) were linked to the recycled Cas12a vector by T4 ligase to generate Cas12a- PDSt1 to Cas12a- PDSt11, and then transformed into E. coli DH5α competent cells. After overnight culture at 37 ℃, single colonies verified by sequencing were inoculated in the LB liquid medium with Kanamycin. After overnight culture at 37 ℃ 220 RPM, plasmids were extracted.
Transcription of sgRNAs in vitro
Specific primers of target sites (Supplemental data set9)were designed, using OsU3p- PDSt1 to OsU3p-PDSt9 plasmids as templates, amplified by high-fidelity enzyme FastPfu and purified by EasyPure PCR Kit. Transcription of purified PCR products in vitro was performed by NEB HiScribe ™ T7 in vitro Transcription Kit. In Vitro Transcription products were purified by TIANGEN RNA Purification Kit.
Protoplast transformation with plasmid or Cas9 RNP complex
The protoplast concentration was adjusted to 2 ×106 – 2 ×107 with MMG, and protoplasts were incubated on ice. 20 μg plasmids were added to 2 ml centrifuge tube, precipitated to the bottom of the tube by centrifugation. 200μl protoplasts was added into the tube, and mixed lightly. 250 μl 50 % PEG 4000 was added and induced transformation for 30 mins in dark. Addition of 900 μl W5 Solution stopped transfection. The sample was centrifuged at 100 g for 3min, and supernatant was discarded. The protoplast was resuspended with 1ml W5 and cultured in dark at 26-28 ℃.
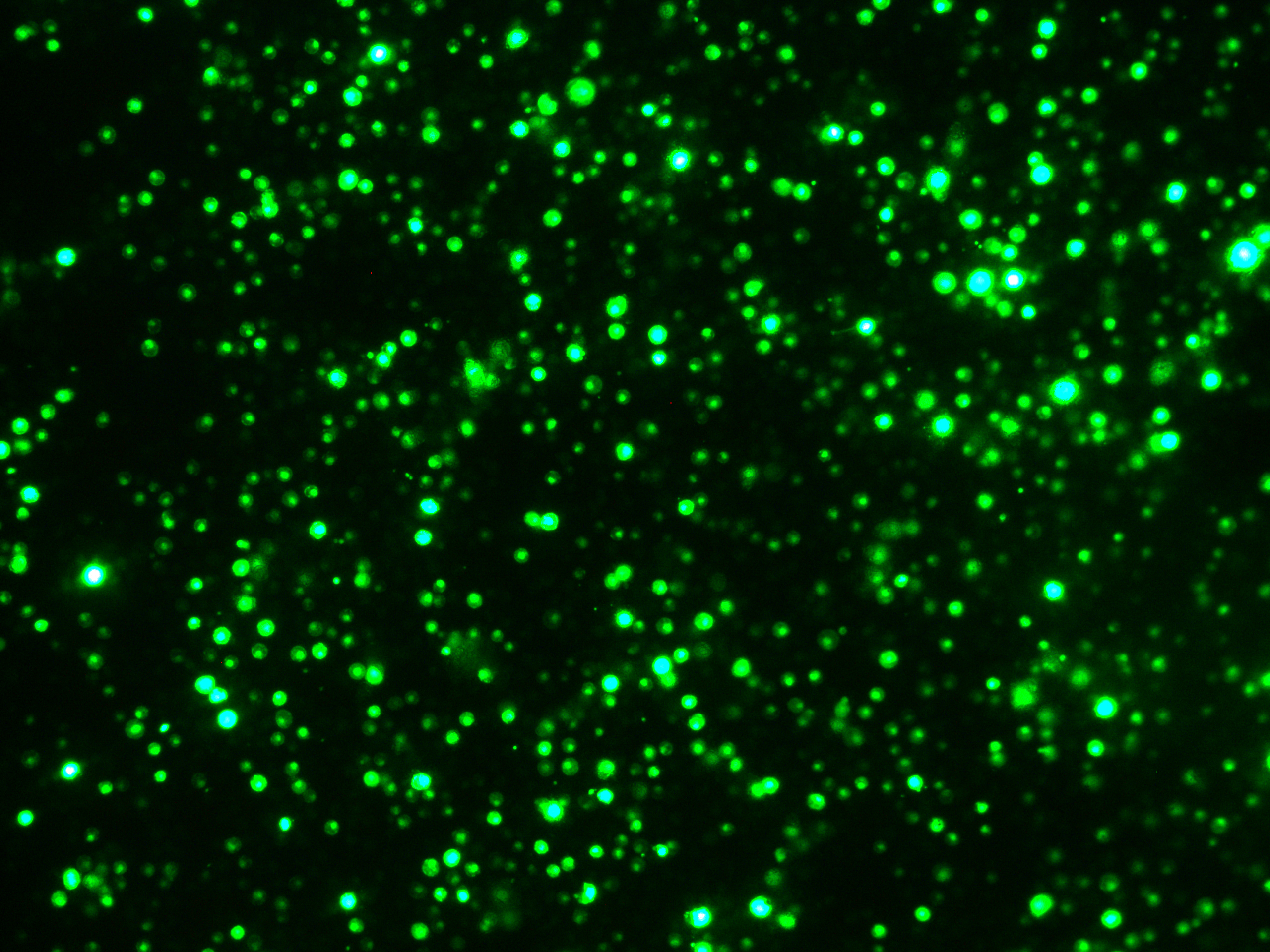
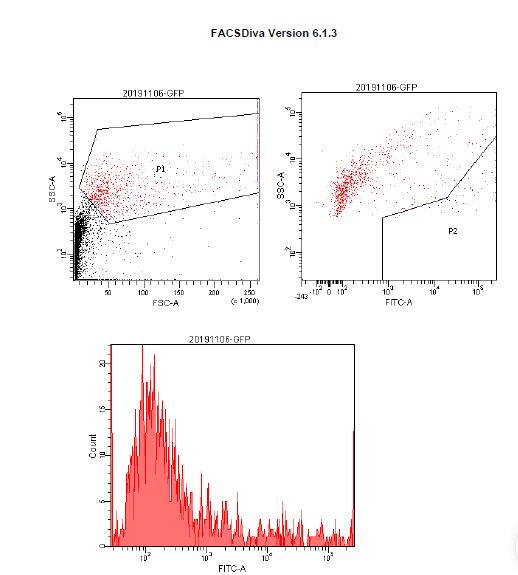
According to the method described above, 10 μg plasmids pUbi-Cas9 was mixed respectively with 10 μg plasmids from OsU3p- PDSt1 to OsU3p- PDSt9, 20 μg plasmids from Cas12a- PDSt1 to Cas12a- PDSt11, Cas9 protein (20 μg) and sgRNA (20 μg). The prepared samples were respectively transformed into banana protoplast by PEG method and then dark cultured. The pUbi-GFP was transformed as a control.
PCR-RE test and monoclonal sequencing
After DNA and RNP transformation, the target sites with specific endonuclease sites were selected for PCR - RE test. Genomic DNA of protoplasts was extracted. The sequence with the length of about 1000 bp containing target sites was amplified by PFU enzyme. The specific PCR products was digested by Eco471 at 37 ℃ for 2 hours and analyzed on 2 % agarose gel by electrophoresis. In order to determine whether there were specific fragments with base mutations, the specific fragments were recovered after electrophoresis, connected to the T-blunt vector, transformed into E. coli DH5α competent cells and then selected for monoclonal sequencing.
RNP cleavage in vitro
DNA fragments containing target 3 and target 4 were amplified by PCR, purified by EasyPure PCR Purification Kit, and eluted by RNase-free water. The cleavage reaction system in vitro was as follows: Cas9 protein (1 μg), sgRNA (1 μg), target fragment (100 ng), 10×Cas9 reaction buffer (20 mM HEPES, pH 7.5, 150 mM KCl, 10 mM MgCl2, 0.5 mM DTT) 2 μl, RNase-free water up to total volume of 20 μl. Samples were incubated at 37 °C for 1 hour and then at 65 °C for 10 mins. Finally samples were tested by electrophoresis on 2 % agarose gel.
Deep amplicon sequencing
The prepared samples of DNA or RNP were transformed into banana protoplast. After 4-5 days dark culture, the banana protoplast were collected by centrifugation at 12000 RPM and then genomic DNA was extracted by TIANGEN DNA extracted Kit. Deep amplicon sequencing primers(Supplemental data set10, Supplemental data set11)were designed and nested PCR was performed to amplify fragments with the length of approximate 200 bp. After gel purification, samples were sent for Deep amplicon sequencing by Shanghai shenggong biology co., Ltd to determine whether there were base mutations in the target sequences and the types of mutations.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}