General methods
All chemicals and solvents were purchased from Fisher Scientific, Acros Organics, Alfa Aesar or Sigma-Aldrich. Solvents were dried by through solvent purification system by passing through activated alumina and copper catalyst columns. All reactions were carried out at room temperature under a nitrogen atmosphere using a nitrogen balloon unless mentioned otherwise. Reactions were monitored by TLC (silica gel, f254) under UV light or by charring (5% H2SO4-MeOH) and the purification was performed by column chromatography on silica gel (230-400 mesh) using the solvent system specified, solvents were used without purification for chromatography. 1H NMR was recorded on Bruker Avance III 600 MHz spectrometer using CDCl3 and D2O as an internal reference. 13C were recorded on Bruker Avance III 600 MHz spectrometer using CDCl3 and D2O as internal reference. High resolution mass spectrometry was recorded on TOF MS-ES+ instrument. Low resolution mass spectrometry was recorded on ESquire-LC-MS.
Metathesis procedure
A solution of dialkene in dichloromethane was degassed by passing nitrogen gas through it for 20 minutes. After that, 1st generation Grubb’s catalyst (10 mol%) was added to the solution and the reaction was kept under nitrogen atmosphere using nitrogen balloon for 7 days or until the catalyst turned dark brown or black. Then, everything was evaporated under reduced pressure and purified using flash column chromatography on silica gel.
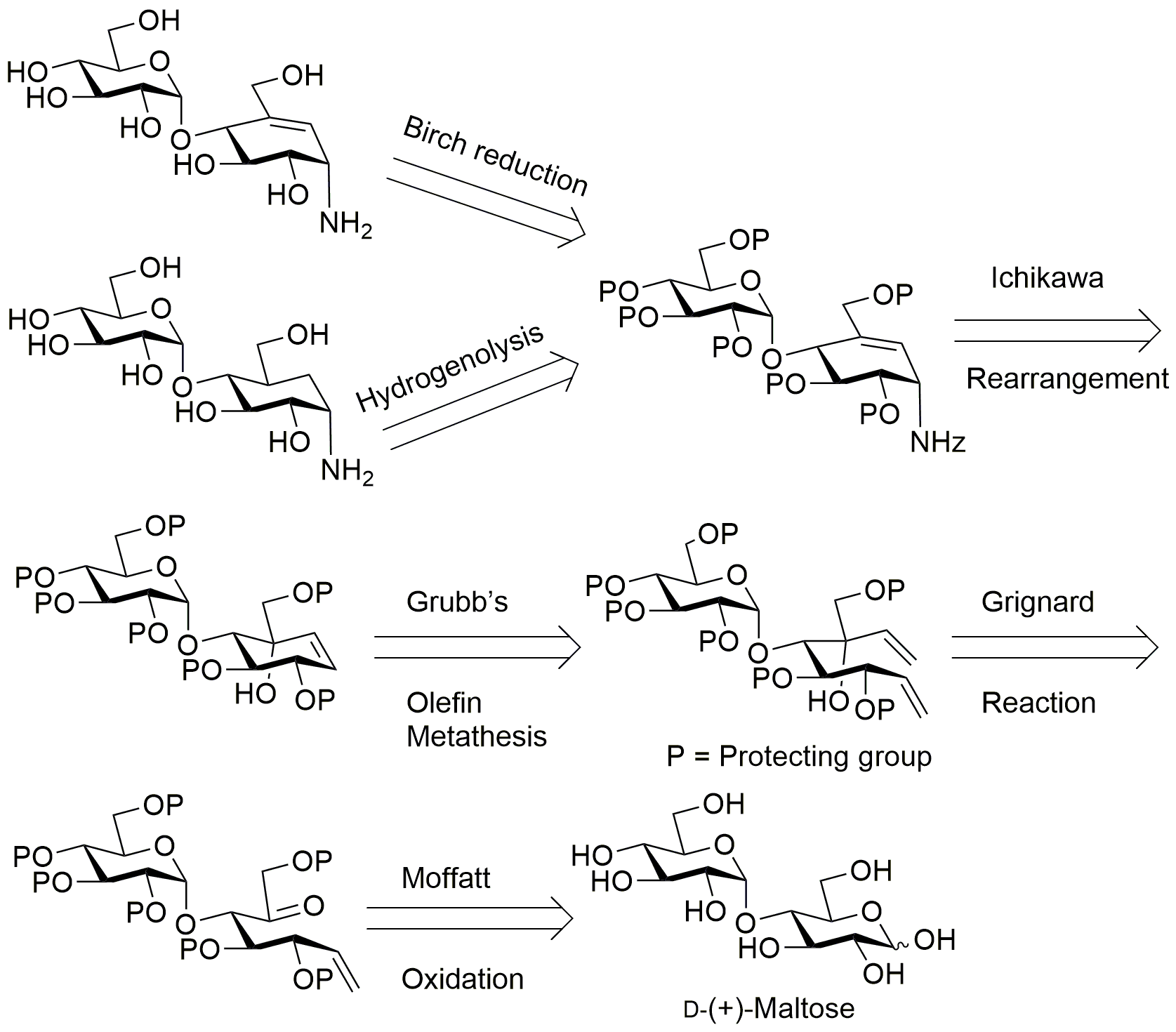
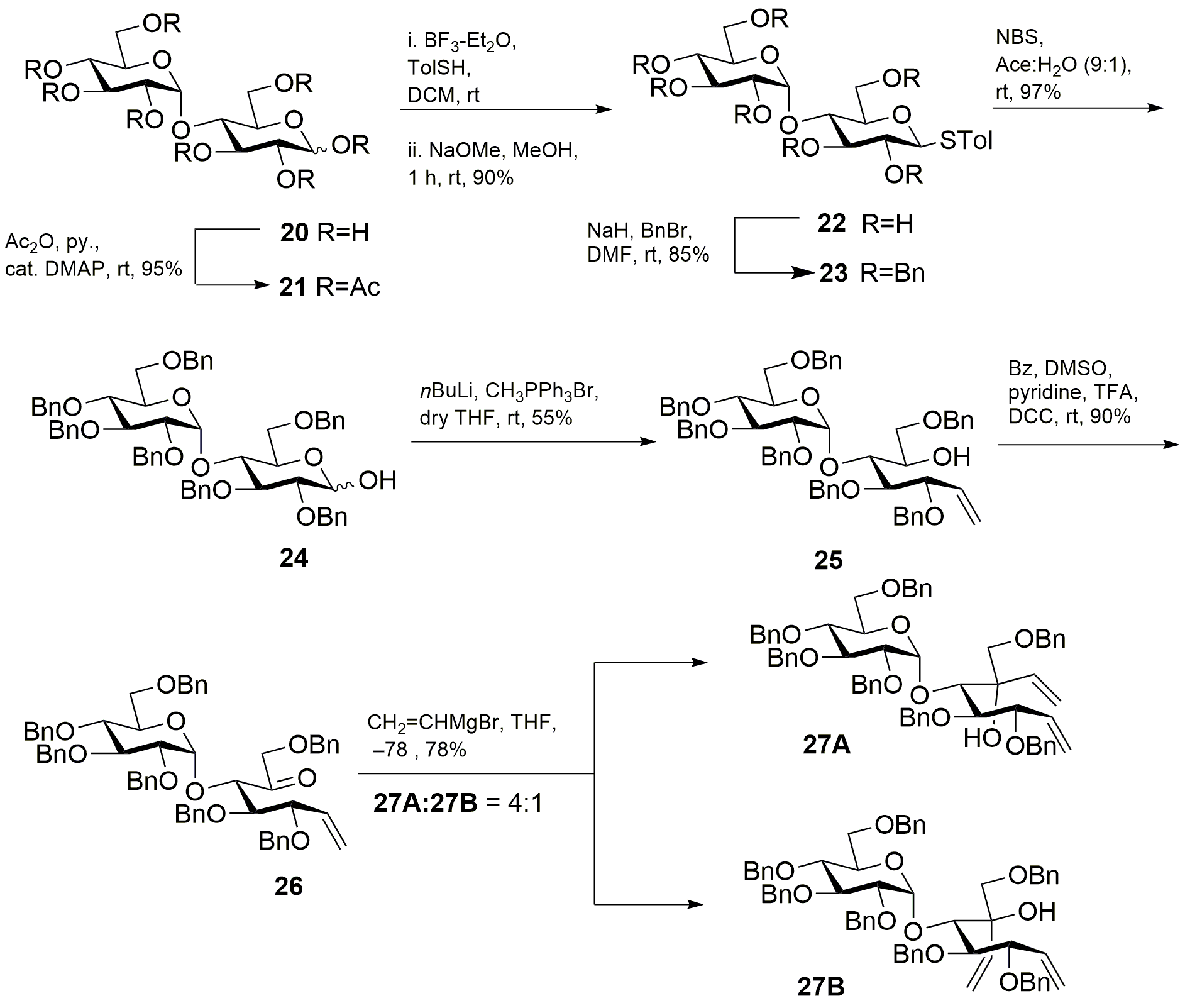
1,2,3,6,2’ ,3’ ,4’ ,6’-octa-O-acetyl-D-maltose (21)65
D-(+)-Maltose monohydrate (2.00 g, 5.84 mmol) was suspended in dry pyridine (7.20 mL, 89.0 mmol) and acetic anhydride (6.28 mL, 66.8 mmol). A catalytic amount of 4-(dimethylamino)pyridine was added to the solution. The solution was stirred at ambient temperature for 16 h. The reaction mixture was diluted with ethyl acetate and washed successively with 1 N HCl (20 mL) and saturated aq. NaHCO3 (40 mL). The resulting organic phase was dried with anhydrous Na2SO4, filtered and the filtrate was concentrated under reduced pressure to give product 21: yield 95% (3.50 g); silica gel TLC Rf = 0.5 (50% ethyl acetate : hexane). All the NMR values match with the reported data. 65
4-methylphenyl 1-thio-β-D-maltopyranopyranoside (22)66
Peracetylated maltose 21 (3.45 g, 5.08 mmol) was dissolved in dichloromethane. 4-Methylthiophenol (1.26 g, 10.2 mmol) and boron trifluoride diethyl etherate (0.75 mL, 6.10 mmol) were added to the solution at 0 °C. The resulting solution was stirred at ambient temperature under nitrogen atmosphere. The reaction was monitored by TLC and appeared to be complete after 4 h. The reaction mixture was diluted with dichloromethane (20 mL) and successively washed with saturated aq. NaHCO3 (25 mL), brine (25 mL), and water (15 mL). The resulting organic phase was dried (anhydrous Na2SO4), filtered and the filtrate was concentrated under reduced pressure. The product was dried overnight and deacetylated by dissolving in dry methanol followed by addition of a catalytic amount of sodium metal until the solution reached pH 9. The reaction was monitored for completion using TLC. The reaction was neutralized by adding Amberlite IRA-118H H+ resin until the pH reached 7. Then, the resin was filtered away and the filtrate was concentrated under reduced pressure to afford the 4-methylthiophenyl maltoside 22: yield 90% (3.10 g). All the NMR values match with the reported data.66
4-methylthiophenyl 2,3,6-Tri-O-benzyl-4-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)-β-D-glucopyranoside (23)67
A solution of 22 (2.95 g, 9.04 mmol) in dry N,N-dimethylformamide (50 mL) was cooled to 0 °C. The solution was treated drop-wise with a suspension of sodium hydride (60% dispersion in mineral oil) (4.55 g, 114.0 mmol) in dry N,N-dimethylformamide. Benzyl bromide (11.6 mL, 95.3 mmol) was added drop-wise over 15 min. and the solution stirred at room temperature for 16 h. The reaction was poured over ice and extracted with ethyl acetate (50 mL). The combined organic layers were washed with brine. The resulting organic phase was dried (anhydrous Na2SO4), filtered and the filtrate was concentrated under reduced pressure to obtain a product. The product was purified by silica gel flash column chromatography. The product fractions were combined, concentrated, and dried in vacuum to afford a yellow oily product 23: yield 85% (2.50 g); silica gel TLC Rf = 0.61 (25% ethyl acetate : hexane). All the NMR values match with the reported data.67
2,3,6-Tri-O-benzyl-4-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)-α/β-D-glucopyranoside (24)44
N-bromosuccinimide (0.90 g, 5.08 mmol) was added to a solution of 6 (2.47 g, 2.45 mmol) in 9:1 acetone-water (50 mL) and stirred at room temperature for 45 min. The solvent was evaporated at room temperature until turbid. A solution of the residue in ethyl acetate (100 mL) was washed successively with saturated aqueous NaHCO3, (3 X 50 mL) and water (3 X 50 mL). The solution was dried with anhydrous Na2SO4 and evaporated. The product was purified by silica gel flash column chromatography. The product fractions were combined, concentrated, and dried in vacuum to afford a yellow oil 24: yield 97% (2.40 g); silica gel TLC Rf = 0.19 (30% ethyl acetate : hexane). All the NMR values match with the reported data.44
3,4,7-Tri-O-benyl-5-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)-D-gluchept-1-enitol (25)45
n-Butyl lithium (2.22 M) in hexanes (5.3 mL, 12.0 mmol) was added drop-wise to a suspension of methyltriphenylphosphonium bromide (3.36 g, 9.44 mmol) in tetrahydrofuran (50 mL) at –20 °C. The solution was stirred at –20 °C for 15 min. and raised to ambient temperature over 1 h. The solution was cooled to –20 °C and compound 24 (2.30 g, 2.36 mmol) dissolved in 50 mL of tetrahydrofuran was added drop-wise. The solution was stirred at –20 °C for 15 min. and allowed to warm to ambient temperature and stirred for an additional 6 h. The solution was diluted with acetone (6 mL) and stirred for 30 min. Diethyl ether (40 mL) was added to precipitate triphenylphosphine oxide. The latter was removed by filtration through Celite™ 545 filter aid. The filtrate was washed successively with satd. aq. NaHCO3 and brine (50 mL X 3). The solution was dried with anhydrous Na2SO4, filtered and the filtrate concentrated under reduced pressure to obtain the crude product. The product was purified by silica gel flash column chromatography on silica gel to give product as a yellow oil 25: yield 55% (1.30 g); Rf = 0.63 (30% ethyl acetate : hexane). All the NMR values match with the reported data.45
3,4,7-Tri-O-benzyl-5-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)-D-gluchept-1-enone (26)
Compound 25 (1.20 g, 1.24 mmol) was dissolved by gentle warming in anhydrous benzene (3.0 mL). To this solution was added dry dimethyl sulfoxide (3.0 mL). To the clear solution was added anhydrous pyridine (99.5 µL, 1.24 mmol), trifluoracetic acid (47.3 µL, 0.62 mmol) and N,N'-dicyclohexylcarbodiimide (0.76 g, 3.70 mmol) in that order. The reaction was left to stir at room temperature for 18 h. After completion of reaction, which was monitored by TLC, benzene (10 mL) was added. The resulting crystalline dicyclohexylurea was removed by filtration and washed with benzene. The combined filtrates and washings were extracted with water (20 mL X 3) to remove dimethyl sulfoxide. The organic layer was dried over anhydrous Na2SO4, evaporated under reduced pressure, and subjected to flash column chromatography on silica gel with 1:6 ethyl acetate–hexane to give product as colorless viscous liquid 26: yield 88% (1.05 g); silica gel TLC Rf = 0.72 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.33–7.23 (m, 33H, aromatic), 7.17–7.15 (m, 2H, aromatic), 5.97 (m, 1H, H-1), 5.24 (m, 2H, =CH2), 4.93 (d, J = 11 Hz, 1H, PhCH), 4.90–4.84 (m, 4H, PhCH , H-1’), 4.68 (dd, J = 12, 4.1 Hz, 2H, PhCH), 4.56 (dd, J = 12, 4.1 Hz, 2H, PhCH), 4.48 (d, J = 10.8 Hz, 1H, PhCH), 4.44–4.37 (m, 4H, PhCH, H-6a), 4,26 (d, J = 6.2 Hz, 1H, H-4), 4.18–4.24 (m, 3H, PhCH, H-6b), 4.09–4.06 (m, 2H, H-3’, H-2), 3.93 (m, 1H, H-5’), 3.85 (dd, J = 6.2, 4.4 Hz, 1H, H-3), 3.79–3.74 (m, 2H, H-4’, H-6a’), 3.54 (dd, J = 9.7, 3.7 Hz, 1H, H-2’), 3.45 (dd, J = 10.8, 1.8 Hz, 1H, H-6b’); 13C NMR (150 MHz, CDCl3): δ 204.40, 138.77, 138.53, 138.28, 138.19, 137.92, 137.91, 137.64, 134.92, 128.41, 128.34, 127.70, 127.63, 118.92, 99.72, 81.73, 80.64, 79.71, 79.71, 79.23, 75.64, 74.90, 74.55, 73.48, 73.20, 73.06, 70.74, 68.06 ppm; mass spectrum (HRMS), m/z = 969.4601 (M+H)+, C62H64O10 requires 969.4578.
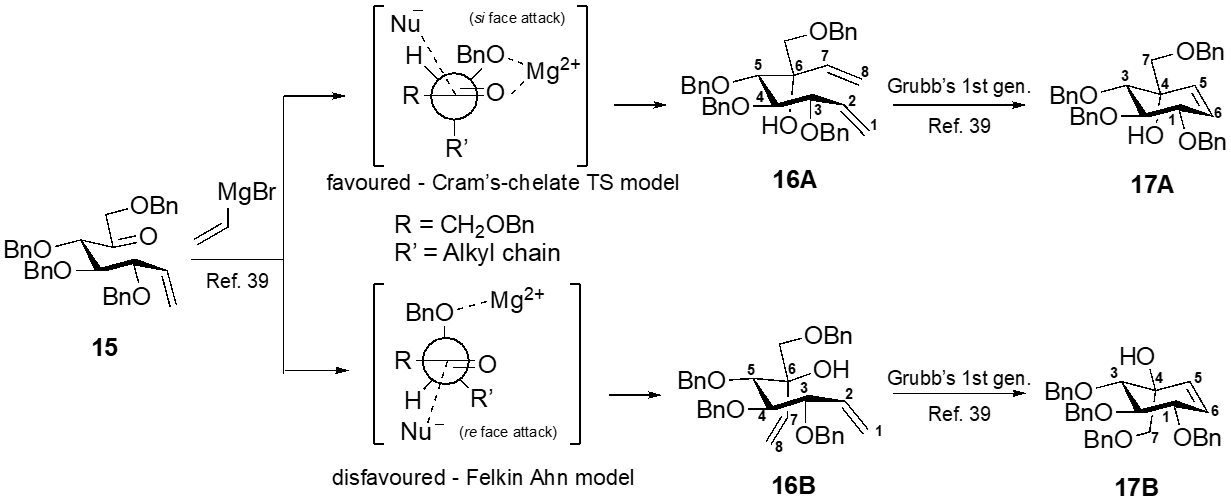
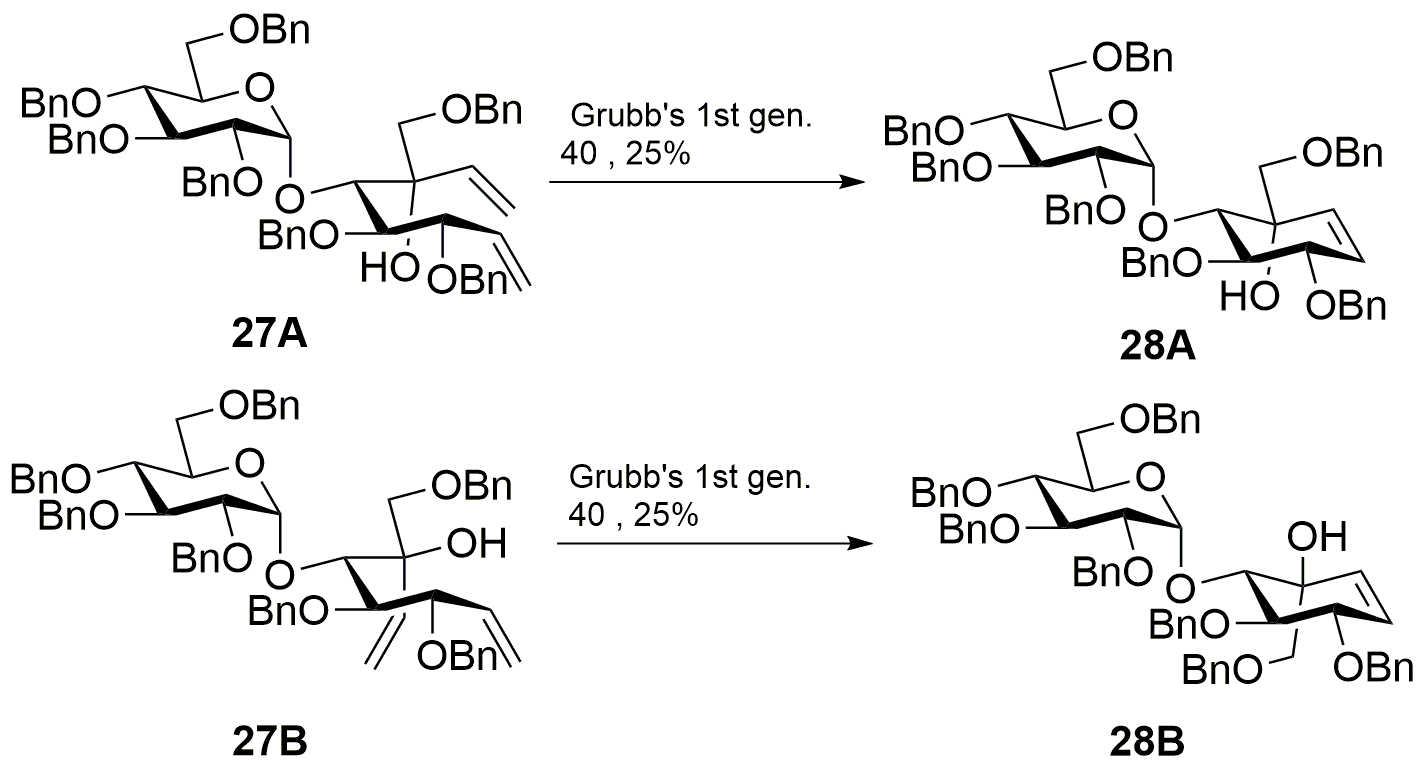
3,4,9-Tri-O-benzyl-5-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)-D-gluco-octa-1,7-dienitol (27A)
To a cooled (–78 °C) solution of 26 (1.04 g, 1.07 mmol) in tetrahydrofuran (15 mL) was added of vinylmagnesium bromide (0.7 M) in tetrahydrofuran (4.57 mL, 3.20 mmol) dropwise. The reaction mixture was stirred for 1 h. at the same temperature. The reaction mixture was warmed to room temperature. Diethyl ether (30 mL) and aq. NH4Cl (30 mL) were added to the reaction mixture. The organic layer was separated, washed with brine (50 mL X 2), and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and purification was performed by flash column chromatography on silica gel with 1:8 ethyl acetate–hexane to afford product as colorless viscous liquid 27A: yield 93% (0.97 g); silica gel TLC Rf = 0.65 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.40–7.23 (m, 31H, aromatic), 7.22–7.17 (m, 4H, aromatic), 6.30 (dd, J = 17.4, 10.6 Hz, 1H, H-7), 5.79 (ddd, J = 17.6, 10.4, 7.5 Hz, 1H, H-2), 5.59 (dd, J = 17.4, 1.7 Hz, 1H, H-8a), 5.37 (br dd, J = 17.3, 1.1 Hz, 1H, H-1a), 5.33 (d, J = 3.4 Hz, 1H, H-1’), 5.27 (ddd, J = 10.2, 6.8, 1.7 Hz, 2H, H-1b, H-8b), 5.01 (d, J = 10.9, 10.9 Hz, 2H, PhCH), 4.89 (d, J = 11.5 Hz, 2H, PhCH), 4.66–4.43 (m, 10H, PhCH, H-3), 4.29 (d, J = 11.7 Hz, 1H, PhCH), 4.11–4.03 (m, 2H, H-5, H-3’), 4.00–3.92 (m, 2H, H-5’, H-4), 3.85 (brs, 1H, OH), 3.76–3.68 (m, 2H, H-4’, H-9a), 3.64 (dd, J = 9.8, 3.5 Hz, 1H, H-2’), 3.55 (dd, J = 10.8, 2.8 Hz, 1H, H-6a’), 3.44 (dd, J = 10.7, 1.6 Hz, 1H, H-6b’), 3.36 (d, J = 8.9 Hz, 1H, H-9b). 13C NMR (150 MHz, CDCl3): δ 139.92, 138.86, 138.71, 138.60, 1338.38, 137.99, 137.97, 137.71, 135.78, 128.35, 128.30, 128.18, 128.11, 127.92, 127.75, 127.65, 127.58, 127.48, 127.43, 127.35, 127.24, 119.35, 1155.67, 97.01, 82.97, 81.77, 79.74, 79.37, 77.61, 77.49, 75.47, 74.93, 74.78, 74.32, 73.44, 73.02, 72.54, 70.77, 70.48, 67.81 ppm; mass spectrum (HRMS), m/z = 997.4899 (M+H)+, C64H68O10 requires 997.4891.
3,4,9-Tri-O-benzyl-5-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)-L-ido-octa-1,7-dienitol (27B)
Flash column chromatography on silica gel with 1:6 ethyl acetate–hexane afforded product as colorless liquid 27B: yield 23% (0.24 g); silica gel TLC Rf = 0.72 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.35–7.13 (m, 33H, aromatic), 7.04 (m, 2H, aromatic), 6.27 (dd, J = 17.3, 10.9 Hz, 1H, H-7), 5.81 (ddd, J = 17.3, 10.2, 8.3 Hz, 1H, H-2), 5.60 (dd, J = 17.4, 1.9 Hz, 1H, H-8a), 5.44 (d, J = 3.4 Hz, 1H, H-1’), 5.37 – 5.26 (m, 3H, H-1a, H-1b, H-8b), 4.98 (d, J = 10.8 Hz, 1H, PhCH), .4.93 (d, J = 11.0 Hz, 1H, PhCH), .4.84 (d, J = 10.8 Hz, 1H, PhCH), 4.80 (d, J = 10.8 Hz, 1H, PhCH), 4.60 (dd, J = 16.5, 11.3 Hz, 2H, PhCH), 4.49–4.33 (m, 8 H, PhCH), 4.25 (t, J = 8.3, 8.3 Hz, 1H, H-3), 4.05 (d, J = 1.8 Hz, 1H, H-5), 3.95–3.90 (m, 2H, H-4, H-3’), 3.84 (d, J = 9.2 Hz, 1H, H-9a), 3.77 (m, 1H, H-5’), 3.69–3.63 (m, 1H, H-4’), 3.54 (dd, J = 9.3, 4.6 Hz, 1H, H-2’), 3.39 (dd, J = 10.6, 2.7 Hz, 1H, H-6a’), 3.33–3.29 (m, 2 H, H-6b’, H-9b), 2.97 (brs, 1H, OH); 13C NMR (150 MHz, CDCl3): δ 139.20, 138.81, 138.68, 138.59, 138.35, 137.93, 137.77, 137.20, 135.86, 128.28, 127.63, 127.54, 127.13, 120.06, 115.69, 94.33, 84.60, 81.93, 79.01, 77.90, 77.30, 78.15, 75.46, 74.46, 74.86, 74.67, 73.43, 72.78, 71.15, 70.44, 67.86 ppm; mass spectrum (HRMS), m/z = 997.4899 (M+H)+, C64H68O10 requires 997.4891.
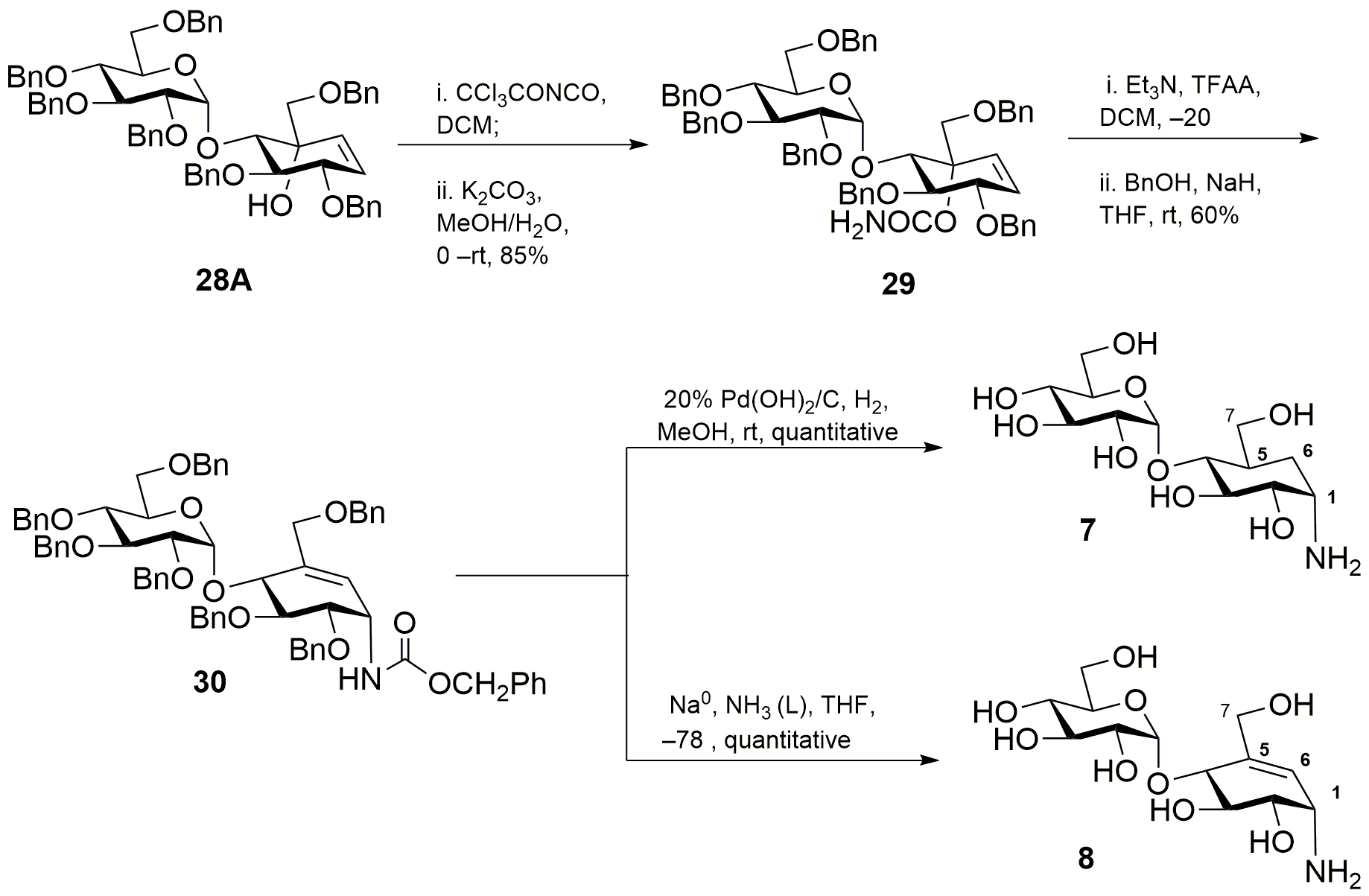
(1D)-(1,3,4/2)-1,2-Di-O-benzyl-4-C-[(benzyloxy)methyl]-3-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)cyclohex-5-ene-1,2,3,4-tetrol (28A)
28A: yield 25% (0.25 g); silica gel TLC Rf = 0.5 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.30–7.20 (m, 33H, aromatic), 7.19–7.11 (m, 2H, aromatic), 5.96 (dd, J = 10.2, 1.9 Hz, 1H, H-6), 5.69 (dd, J = 10.3, 1.8 Hz, 1H, H-5), 5.63 (d, J = 3.7 Hz, 1H, H-1’), 4.98 (d, J = 11.7 Hz, 1H, PhCH), 4.91–4.81 (m, 3H, PhCH), 4.77 (d, J = 10.9 Hz, 1H, PhCH), 4.67 (dd, J = 29.4, 11.7 Hz, 2H, PhCH), 4.59–4.43 (m, 6H, PhCH), 4.36 (d, J = 12.2 Hz, 1H, PhCH), 4.22 (m, 1H, H-1), 4.21–4.17 (m, 2H, H-2, H-3), 3.97 (t, J = 9.3 Hz, 1H, H-3’), 3.89 (m, 1H, H-5’), 3.70–3.63 (m, 2H, H-4’, H-7b), 3.56 (ddd, J = 13.5, 10.2, 3.5 Hz, 2H, H-2’, H-6a’), 3.49(dd, J = 10.5, 1.8 Hz, 1H, H-6b’), 3.39 (d, J = 9.1 Hz, 1H, H-7a), 3.22 (brs, 1H, OH); 13C NMR (150 MHz, CDCl3): δ 139.10, 138.66, 138.34, 138.31, 138.10, 137.96, 137.78, 130.75, 130.31, 128.35, 128.33, 128.27, 128.17, 128.08, 127.92, 127.84, 127.77, 127.65, 127.55, 127.41, 127.66, 126.92, 97.02, 81.77, 80.82, 80.10, 79.60, 77.54, 75.48, 74.95, 74.12, 73.49, 73.10, 72.98. 72.74, 71.50, 68.10 ppm; mass spectrum (HRMS), m/z = 991.4418 (M+Na)+, C62H64O10 requires 991.4397.
(1D)-(1,3,4/2)-1,2-Di-O-benzyl-4-C-[(benzyloxy)methyl]-3-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)cyclohex-5-ene-1,2,3,4-tetrol (28B)
28B: yield 25% (60.0 mg); silica gel TLC Rf = 0.5 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.35–7.15 (m, 35H, aromatic), 5.74 (ddd, J = 22.2, 10.4, 1.9 Hz, 2H, H-5, H-6), 5.32 (d, J = 3.8 Hz, 1H, H-1’), 5.04 (d, J = 12.0 Hz, 1H, PhCH), 4.93 (d, J = 10.9 Hz, 1H, PhCH), 4.87–4.76 (m, 3H, PhCH), 4.42–4.64 (m, 9H, PhCH), 4.27 (dt, J = 7.0, 1.8 Hz, 1H, H-1), 4.16 (dd, J = 10.5, 7.1 Hz, 1H, H-2), 3.95–3.89 (m, 2H, H-3, H-3’), 4.93 (d, J = 10.9 Hz, 1H, PhCH), 4.93 (d, J = 10.9 Hz, 1H, PhCH), 3.73 (d, J = 9.4 Hz, 1H, H-7a), 3.67 (d, J = 9.4 Hz, 1H, H-7b), 3.61–3.51 (m, 4H, H-2’, H-4’, H-6a’, H-6b’), 3.40 (brs, 1H, OH); 13C NMR (150 MHz, CDCl3): δ 139.37, 138.72, 136.64, 136.21, 138.13, 137.99, 137.80, 132,14, 128.41, 128.38, 128.37, 128.32, 128.17, 128.10, 128.06, 128.03, 127.90, 127.80,, 127.76, 127.73, 127.62, 127.56, 127.47, 126.99, 126.95, 126.66, 99.37, 84.31, 81.67, 80.95, 80.46, 79.24, 77.24, 75.52, 75.12, 75.05, 74.37, 73.62, 73.58, 73.53, 72.41, 71.33, 71.11, 68.51, 29.73 ppm; mass spectrum (HRMS), m/z = 991.396 (M+Na)+, C62H64O10 requires 991.4397.
(1D)-(1,3,4/2)-1,2-Di-O-benzyl-4-C-[(benzyloxy)methyl]-4-O-carbamoyl-3-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl)cyclohex-5-ene-1,2,3,4-tetrol (29)
A solution of 28A (0.24 g, 0.25 mmol) in dichloromethane (10 mL) was cooled at 0 °C and treated dropwise with trichloroacetyl isocyanate (60 µL, 0.50 mmol). The reaction mixture was stirred for 30 min. at the same temperature and evaporated. The residue was dissolved in methanol (10 ml) and water (1 ml), was cooled to 0 °C and treated with potassium carbonate (0.07 g, 0.49 mmol). The reaction mixture was stirred for 2 h. at that temperature and warmed to room temperature and again stirred for another 2 h. After completion of the reaction, methanol was evaporated and the aq. solution. was diluted with water (10 mL) and extracted with dichloromethane (20 mL X 3). The organic layer was separated washed with brine (10 mL X 3), dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and purification was performed by flash column chromatography on silica gel with 1:3 ethyl acetate–hexane to afford product as colorless liquid 29: yield 85% (0.21 g); silica gel TLC Rf = 0.25 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.36–7.20 (m, 33H, aromatic), 7.16–7.14 (m, 2H, aromatic), 6.32 (dd, J = 10.2, 1.9 Hz, 1H, H-6), 6.01 (dd, J = 10.3, 1.8 Hz, 1H, H-5), 5.53 (d, J = 3.7 Hz, 1H, H-1’), 5.07 (d, J = 11.7 Hz, 1H, PhCH), 4.92 (m, 2H, PhCH), 4.86 (d, J = 11.0 Hz, 1H, PhCH), 4.65 (dd, J = 11.4, 9.0 Hz, 2H, PhCH), 4.59–4.56 (m, 2H, PhCH), 4.52–4.45 (m, 4H, 2 PhCH), 4.35 (d, J = 12.0 Hz, 1H, PhCH) 4.28–4.23 (m, 3H, H-1, H-2, H-3), 4.14–4.08 (m, 3H, H-3’, H-7a, H-7b), 4.02 (m, 1H, H-5’), 3.68 (t, J = 9.6, 9.6 Hz, 1H, H-4’), 3.61 (dd, J = 10.2, 3.0 Hz, 1H, H-6a’), 3.54 (dd, J = 9.6, 3.6 Hz, 1H, H-2’), 3.50 (d, J = 10.2 Hz, 1H, H-6b’). 13C NMR (150 MHz, CDCl3): δ 155.39, 139.32, 138.84, 138.30, 138.24, 138.17, 138.13, 137.87, 130.68, 129.67, 128.41, 128.39, 128.36, 128.31, 128.25, 128.19, 128.12, 128.05, 127.99, 127.93, 127.90, 127.77, 127.69, 127.57, 127.53, 127.39, 127.07, 126.97, 98.13, 81.53, 81.09, 80.50, 79.77, 79.75, 77.77, 75.93, 75.47, 75.16, 74.43, 73.47, 73.16, 72.50. 71.76, 70.90, 69.78, 68.51 ppm; mass spectrum (HRMS), m/z = 1034.4463 (M+Na)+, C63H65NO11 requires 1034.4455.
(1D)-(1,3,4/2)-1,2-Di-O-benzyl-6-[(benzyloxycarbonyl) amino]-4-[(benzyloxy)methyl]-3-O-(2’,3’,4’,6’-tetra-O-benzyl-α-D-glucopyranosyl) cyclohex-4-ene-1,2,3-triol (30)
Trifluoracetic anhydride (56.0 µL, 0.39 mmol) was added to a solution of carbamate 29 (0.20 g, 0.19 mmol ) and triethylamine (165 µL, 1.19 mmol) in dry tetrahydrofuran (10 mL) and was cooled to 0 °C. The resulting mixture was slowly warmed to room temperature and stirred for 1 h. In a separate flask, sodium hydride suspension in 60% mineral oil (0.01 g, 0.39 mmol) was added to a solution of benzyl alcohol (41.0 µL, 0.39 mmol) in dry tetrahydrofuran (2 mL) at 0 °C. After 1 hr, the solution of sodium benzyloxide was added to the generated isocyanate and the reaction progress was followed by TLC. After 24 h. volatiles are removed under reduced pressure. The reaction mixture was diluted with water (10 mL) and extracted with dichloromethane (10 mL X 4). The organic layer was separated washed with brine (10 mL X 2), dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and purification was performed by flash column chromatography on silica gel with 1:5 ethyl acetate–hexane to afford product as colorless liquid 30: yield 60% (0.13 g); silica gel TLC Rf = 0.43 (30% ethyl acetate : hexane); 1H NMR (600 MHz, CDCl3) δ 7.23–7.11 (m, 40H, aromatic), 5.92 (d, J = 3.0 Hz, 1H, H-5), 5.49 (d, J = 9.7 Hz, 1H, NH), 5.10–5.05 (m, 3H, H-1’, cbz), 4.79–4.71 (m, 3H, H-6, PhCH), 4.68–4.57 (m, 6H, PhCH), 4.51–4.36 (m, 6H, PhCH), 4.29 (br d, J = 4.8 Hz, 1H, H-3), 4.25 (d, J = 12.6 Hz, 1H, H-7a), 4.04 (dd, J = 4.8, 1.9 Hz, 1H, H-2), 3.94 (d, J = 12.6 Hz, 1H, H-7b), 3.90-3.87 (m, 2H, H-3’, H-5’), 3.76 (t, J = 4.8, 4.8 Hz, 1H, H-1), 3.68 (t, J = 9.6 Hz, 1H, H-4’), 3.63 (dd, J = 10.6, 2.9 Hz, 1H, H-6a’), 3.56 (dd, J = 9.8, 3.6 Hz, 1H, H-2’), 3.47 (dd, J = 10.6, 1.9 Hz, 1H, H-6b’). 13C NMR (150 MHz, CDCl3): δ 156.23, 138.71, 138.41, 138.23, 138.01, 137.93, 137.92, 136.65, 135.29, 128.49, 128.46, 128.41, 128.36, 128.34, 128.28, 127.99, 127.98, 127.92, 127.86, 127.74, 127.69, 127.59, 127.52, 127.49, 127.35, 97.58, 81.98, 79.52, 77.67, 75.93, 75.61, 75.33, 74.91, 73.96, 73.49, 73.03, 71.81, 71.77, 71.26, 71.03, 70.95, 68.16, 66.61, 46.54 ppm; mass spectrum (HRMS), m/z = 1124.4922 (M+Na)+, C70H71NO11 requires 1124.4925.
L-chiro-Inisitol-1-amino-1,5,6-trideoxy-4-O-(α-D-glucopyranosyl)-5-(hydroxymethyl) or 4-α-glycoside derivative of validamine (7)
Compound 30 (40.0 mg, 0.04 mmol) was dissolved in 10 mL of methanol and 20% Pd(OH)2/C on carbon (20.0 mg) was added. The mixture was stirred for 16 h. under hydrogen (1 atm.). The catalyst was filtered away through a plug of Celite® 545 and washed with methanol (5mL). The combined filtrate and washings were concentrated to dryness and the residue was passed through reverse phase C-18 silica gel (washed with water) to afford 7 as a white solid: yield quantitative (5.00 mg); 1H NMR (600 MHz, D2O) δ 5.09 (d, J = 3.84 Hz, 1 H), 4.21 (m, 1 H), 3.92 (m, 2 H), 3.75–3.52 (m, 8 H), 3.35–3.32 (m, 1H), 2.13 (m, 1H), 1.71–1.55 (m, 2H). 13C NMR (151 MHz, D2O): δ 96.30, 73.06, 72.87, 72.69, 70.93, 69.04 (2C), 66.14, 62.37, 60.25, 48.94, 36.62, 21.63 ppm; mass spectrum (HRMS), m/z = 340.1599 (M+H)+, C13H25NO9 requires 340.1600.
α-D-Glucopyranoside, 4-amino-5,6-dihydroxy-2-(hydroxymethyl)-2-cyclohexen-1-yl or 4-α-glycoside derivative of valienamine (8)
Ammonia was condensed into a solution of 30 (60.0 mg, 0.05 mmol) in tetrahydrofuran (5 ml) using a dry ice cooled cold finger apparatus. The solution was treated with sodium in small pieces, until a blue color in the solution persisted. After stirring for 10 min. at –78 °C temperature the mixture was treated with NH4Cl (100 mg), stirred at room temperature overnight and evaporated. The residue was extracted with methanol, filtered, and evaporated. The residue (20.0 mg) was adsorbed on 3 mL of neutral Dowex 50-WX-8 (washed with water). After washing with water (10 mL), elution with 2% aqueous ammonia gave 8 as a white solid: yield quantitative (5.00 mg); 1H NMR (600 MHz, D2O): δ 5.71 (d, J = 4.2 Hz, 1H), 5.26 (d, J = 3.9 Hz, 1H), 4.16–4.06 (m, 3H), 3.93–3.85 (m, 3H), 3.71–3.58 (m, 3H), 3.50–3.40 (m, 2H), 3.27–3.23 (m, 1H). 13C NMR (151 MHz, D2O): δ 142.85, 117.37, 97.76, 75.27, 72.65, 72.63, 71.09, 70.14, 69.17, 66.40, 61.34, 60.31, 48.57 ppm; mass spectrum (HRMS), m/z = 338.1440 (M+H)+, C13H23NO9 requires 338.1446.
Protein purification
The expression construct of Sco GlgE1-V279S (pET32-Sco GlgE1-V279S),27 that encodes for a non-cleavable C-terminal polyhistidine tagged protein, was used to transform T7 express E. coli cells. The large-scale cultures were grown at 37 °C in LB media with 264 mM concentration of carbenicillin. When O.D 600nm reached 0.6, cultures were cooled to 16 °C and cells were induced by the addition of 1 mM Isopropyl β-D-1-thiogalactopyranoside. After 16 hours of induction, cells were harvested by centrifugation and resuspended in a lysis buffer consisting of 20 mM Tris pH 7.5, 500 mM NaCl, 10 % glycerol, 0.5 mM imidazole and 0.3 mM tris(2-carboxyethyl) phosphine (TECP). The cell suspension was incubated on ice with lysozyme and DNase I for 30 min and lysed by sonication. The resulting lysate was clarified by centrifugation and subjected to a 5 mL metal affinity cobalt column that had been equilibrated with lysis buffer. The unbound protein was washed by passing 25 column volumes of lysis buffer through the column and Sco GlgE1-V279S was eluted isocratically with the elution buffer of 20 mM Tris pH 7.5, 500 mM NaCl, 150 mM imidazole and 0.3 mM TCEP. The excess salt and imidazole were removed from Sco GlgE1-V279S by overnight dialysis against a buffer consisting of 20 mM Tris pH 7.5, 150 mM NaCl and 0.3 mM TCEP.
Inhibition studies
The inhibition studies for 7 and 8 were performed separately by using EnzChek Phosphate Assay Kit. Prior to the assay, stock solutions of 1 mM MESG, 20 mM concentrations of 7 and 8 (dissolved dH2O) were prepared and stored at -20 °C. In the assay, all the reactions were carried out in a 96-well format, 50 µL reaction volume at 25 °C for 20 minutes in a continuous manner. The reactions were initiated by addition of 50 nM Sco GlgE1-V279S to the reaction mixture consisting of 20 mM Tris pH 7.5, 150 mM NaCl, 500 nM MESG, 0.25 U PNP, 0.5 mM glycogen (as maltose acceptor) and 250 µM M1P.27,52 In the various reactions, the compound 8 concentration was varied from 0 to 1000 µM, except in the positive control that lacked inhibitor, and the negative control that lacked enzyme. All of the reactions were performed in triplicate using a Synergy H4 plate reader (Bio Tek) to measure the absorbance at 360 nm. The percent enzymatic activity was calculated by using the equation (V/V0) × 100, where V and V0 denote the rates of the inhibited and uninhibited enzyme (positive control), respectively. The percent enzymatic activity vs inhibitor concentration was plotted using Prism 8 and the equation y = 100/ (1+( IC50/x) Hillslope) was used to determine the IC50. Using this assay, 100 µM concentration of both 7 and 8 were tested separately against Sco GlgE1-V279S. Dose-response experiments for 8 were performed as described above while varying the concentration of 8.
Crystallization of Sco GlgE1-V279S complexes
Prior to the experiment, 20 mM aqueous solutions of 7 and 8 were prepared. The protein-compound mixture was prepared by mixing the Sco GlgE1-V279S with each compound separately to a final concentration as 8 mg/mL51 of Sco GlgE1-V279S and 10 mM of the respective compound. The hanging drop vapor diffusion method was used for the crystallization and each crystallization drop consisted with 2 µL of Sco GlgE1-V279S/compound mixture and 2 µL of the well solution (0.2 mM Sodium citrate pH 7 and 10 % PEG 3,350).45 The crystallization drop was equilibrated against 100 µL of the well solution and crystals were formed within 3 days. Following crystal formation, an additional 4 µL of the appropriate 20 mM compound stock was added to the drop and incubated for an additional 12 hours. Crystals were cryoprotected by adding 4 µL of 50% PEG 2000 to the crystallization drop, and crystals were flash-cooled using liquid nitrogen in preparation for the cryo-crystallographic experiments.
X-ray Diffraction experiments
The LS-CAT beamline at Advanced Photon Source of Argonne National Labs, IL was used to perform the X-ray diffraction experiments. The X-ray diffraction data corresponding to the Sco GlgE1-V279S/7 and Sco GlgE1-V279S/8 complexes were indexed and reduced, using DIALS, iMosflm, and Scala in the CCP4 suite. The refinements were carried out by using PHENIX, and the previously published Sco GlgE1-V279S in complex with maltose-C-phosphonate (RCSB accession number 4U31) was used for difference Fourier analysis to calculate phases and difference maps prior to manual and computational refinement.68 The visualization and manual refinements of the structures were carried out using COOT.69 The final occupancy of each ligand was changed manually and then refined in PHENIX.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}