1. Characterization of TiO2NPs and MWCNTs
Titanium dioxide nanomaterials (TiO2NPs, anatase, nanopowder, ≥ 99.7%, Product number 637254, Batch number MKCK4358) and multi-walled carbon nanotubes (MWCNTs nanopowder, ≥ 98%, Product number 698849, Batch number MKBH5811V) were purchased from Sigma-Aldrich (USA). The nanomaterials size was examined by Tecnai G2 F30 field emission transmission electron microscopy (TEM) (FEI, USA) and quantified by Nano Measure 1.2. Endotoxin was measured by the endpoint chromogenic Limulus Amebocyte lysate (LAL) assay kit (BIOENDO, China).
2. Preparation of Nanomaterials
CoNPs, TiO2NPs and MWCNTs were diluted with ddH2O to a final concentration of 1mg/mL in 1.5 mL microtubes, respectively. Before applying them to the cells, the solution was sonicated in a bath-type sonicator (KQ-500E, Kunshan Ultrasonic Instruments Co.LTD, China) for 10 min and shaken every three mins.
3. Cell culture and nanomaterial exposure
Human glioma cells, U251, were purchased from the State Key Laboratory of Genetic Resources and Evolution (Yunnan, China). U251 is a commonly model to study neurotoxicity 21,22. U251 were cultured in 1640 medium supplemented with 10% fetal bovine serum (FBS) and 100 units/mL of penicillin−streptomycin. Cells were cultured at 37 °C as monolayers in a humidified atmosphere containing 5% CO2.
When cell density reached 70−80% confluence, the medium was changed to 1640 without FBS. And then the cells were treated with various concentrations of CoNPs, TiO2NPs and MWCNTs for 24 h to wait for the next measurement.
In order to select the appropriate concentration, we measured the cell viability of U251 cells by exposing them to a series of concentrations of nanomaterials. We have then selected the concentration with a similar degree of cell damage (30 μg/mL) as the exposure dose for the following study (Additional Figure 1).
4. Primary astrocyte culture and exposure
Mice were housed in stainless steel cages in a ventilated animal facility at a temperature maintained in 22 ± 2 °C and relative humidity of 65 ± 10% under a 12 h light/dark cycle, and feed with sterilized food and distilled water. All mice were treated humanely throughout the experimental period.
The newborn puppies (within 24 h) from C57BL/6 were sacrificed through inhaling carbon dioxide. And the cortex was isolated from mice and the meninges and blood vessels were removed in Hank's equilibrium salt solution. Next, the cortex was transferred to the F12 medium with 2.5% trypsin to digest at 37°C for 30 min after mincing. After centrifugation and suspension, the mixed glial cells were plated on a T-25 flask coated with poly-lysine and cultured with DMEM medium containing 10% fetal bovine serum. The cells were cultured at 37°C in an atmosphere of 5% CO2 and 95% air. Cell culture medium was replaced 24 h after plating and every 2 days. After 7 to 10 days, astrocyte was shaken at 250 RPM for 14 h to remove unwanted cells, including microglia, neurons, and fibroblasts. It was then digested with 2.5% trypsin at 37℃ for 5 min and seeded in 12 well-plate for the following measurement.
To validate the purity of PA, immunofluorescent was used. Briefly, 4% w/v paraformaldehyde was added12 well-plate and incubated at 4°C for 15 min. Then cells were permeabilized for 15 min with 0.15% Triton X-100 in phosphate-buffered saline (PBS) and blocked by 10% normal goat serum (NGS) for 1 h at room temperature (RT). For GFAP staining, cells were incubated with anti-GFAP antibody (1:500) (Abcam, Cambridge, England) at 4°C overnight. And then Alexa Fluora 488 conjugated secondary antibody was incubated at RT for 1 h, and DAPI (Beyotime, Shanghai, China) was used for nuclear staining. The purity of PA (%) = GFAP positive cells / DAPI positive cells × 100 %. 150 cell was calculated in each well. The purity of PA is over 95% (Additional Figure 2).
When PA density reached 70−80% confluence, the medium was changed to DMEM without FBS. And then PA was exposed to CoNPs, TiO2NPs and MWCNTs for 24 h to wait for the next measurement.
5. Assessment of cell viability
Cells were seeded in a 96-well plate with a density of 5 × 103 /mL in each well, added with cell medium (each well 100 μL) and exposed to nanomaterials for 24 h. And then CCK-8 reagent (Beyotime, Shanghai, China) was added to 96-well and incubated at 37℃ for 1 h. A microplate reader (Multiskan FC, Thermo Fisher Scientific, Waltham, MA, USA) was adopted to measure the absorbance (A) at 450 nm. Six parallel wells were set in each group, and the mean value was obtained. Cell survival rate was calculated using the following formula: cell survival rate (%) = (Absorbance of the experimental group/ Absorbance of the control group) × 100%.
6. High content screening system (HCS)
Cells were seeded into the 24-well plates with a density of 2 × 103 /mL in each well. After exposure to nanomaterials, plates were observed in high content screening system (Perkinelmer, Massachusetts, USA) for 24 h and captured the picture every 15 min.
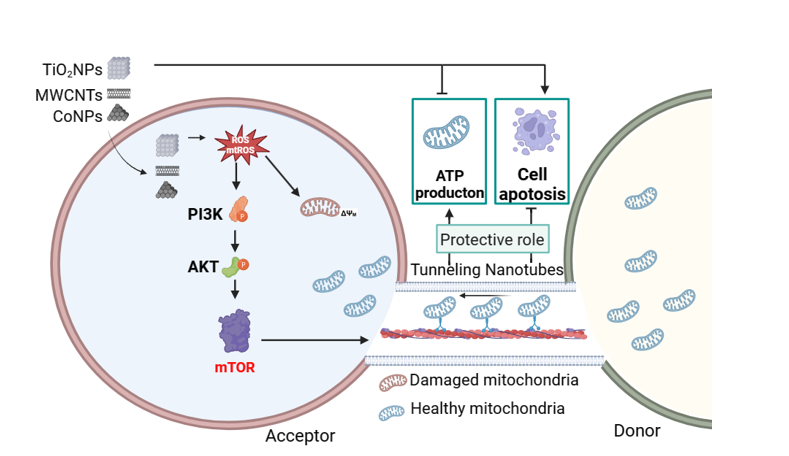
7. Quantification of the numbers of TNTs and mitochondrial transfer
Using TCS SP5 confocal microscope (Leica, Germany), fields of sub-confluent cells were randomly taken with a 20X objective. At least ten images were taken for each experimental group. The number of TNTs per 100 cells was calculated for TNTs in U251 cells using Image J. At least fifty TNTs were imaged in each group. In each field, the percentage of TNTs containing mitochondria was quantified.
8. ATP plasmid transfection
Cells were seeded in the 12-well plates at a density of 50 % for 24 h and transferred with pCMV-Mito-AT1.03 plasmid (Beyotime, Shanghai, China) using the lipo8000 (Beyotime, Shanghai, China) according to the manufacturer’s instruction. Afterwards, the transfected cells were exposed to nanomaterials. The intensity of ATP was captured by fluorescence microscope and quantified by Image J 2.1.
9. Uptake of three types of nanomaterials
The uptake of nanomaterials can be assessed using flow cytometry, as reported by Suzuki et al 23. Cells treated with nanomaterials were washed 3 times with PBS to remove free particles. The cells were re-suspended in DMEM, and the number of particles taken up was analyzed with flow cytometry (FACSCanto II, Becton Dickinson, Franklin Lakes, NJ). A profile of the sample can be obtained by examining both the forward-scattered light (FSC) and the side-scattered light (SSC). As each cell intercepts the path of the laser beam, the light that passes around the cell is measured as FS, indicating cell size. The light scattered at a 90° angle to the axis of the laser beam is measured as SS and is related to intracellular density. Thus, changes in cellular SS, after treatment with nanomaterials, can report upon the uptake potential of the nanomaterials.
10. Detection of mitochondrial reactive oxygen species (mtROS) and reactive oxygen species (ROS)
The mtROS and ROS of treated cells were measured by Mito-SOX (Invitrogen, Carlsbad, USA) and DCFH-DA dye staining (Beyotime, Shanghai, China), respectively. Briefly, cells were exposed to nanomaterials for 24h, and then cells were incubated with 0.5 μM Mito-Sox or 1 μM of DCFH-DA for half an hour at 37°C. Finally, the mtROS and ROS were measured by fluorescence microscope.
11. Measurement of mitochondrial membrane potential (MMP)
The MMP of cells was detected by JC-1 probe (Beyotime, Shanghai, China). Briefly, cells were exposed to nanomaterials for 24 h, and then cells were incubated with 1 μM of JC-1 probe for 0.5 h at 37°C. Finally, the MMP was measured by fluorescence microscope, and quantified by Image J according to literature 24.
12. Western blot
Exposed cells were washed three times with cold PBS and collected and then lysed with 120 μL ice-cold RIPA lysis buffer (Beyotime, Shanghai, China). Afterward, cell-free supernatants were obtained by centrifugation of the lysates at 12 000 g for 25 min at 4 °C. Sodium dodecyl sulfate (SDS) loading buffer was added to each supernatant, and subsequently boiled for 10 min to generate SDS-PAGE samples. 20 μL samples were electrophoresed on a 10% SDS polyacrylamide gel. Proteins were then transferred onto a PVDF membrane. After blocking the membrane with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween-20 for 1 h at 25 °C, the blots were incubated with primary antibodies of interest overnight at 4 C. After washing with TBST for 5 times, the blots were incubated with peroxidase-conjugated secondary antibody. The binding of antibodies was detected by chemiluminescence staining using the ECL detection kit (Amersham). The grayscale of protein bands was analyzed by Image J software. Primary antibodies were used at the following concentrations: p-mTOR (1:2000), mTOR (1:2000), P110 (1:2000), P85 (1:2000), p-PI3K (1:2000), AKT (1:2000), p-AKT (1:2000), beta-ACTIN (1:3000), anti-rabbit peroxidase-conjugated secondary antibodies (1:10000).
13. Statistical analysis
Data were analyzed with SPSS software, version 19.0 (IBM Corporation, Armonk, NY, USA). One-way analysis of variance (ANOVA) was used for multiple comparisons. The experimental data with heterogeneity variance were analyzed by using Kruskal–Wallis nonparametric test among different dosage groups. P value <0.05 indicates statistical significance. All the experiments were carried out in independent triplicates and three individual experiments were performed unless otherwise specified.
{kind=link}