SARS-CoV-2 is a novel coronavirus strain and it is new to the scientific communities. Therefore, in-depth monitoring of all aspects of the COVID-19 has been started. For example, signs and symptoms [38], mode of transmission [39], WHO-solidarity trials [40], contact tracing by mobiles apps such as “Arogya Setu” by India [41], CRISPR-Cas based rapid diagnostic of SARS-CoV-2 [42], and also monitoring daily cases by crowd-sourcing from https://www.covid19india.org/. Moreover, several reports are coming out as preprints and formal peer-review publications about repurposing known drugs [2] and antiviral drugs against the SARS-CoV-2 [43]. Reports suggesting possible anti-COVID-19 effects of different phytochemicals by in silico screening methods [4]. Indeed to address the urgent need for a safe and efficacious vaccine against the SARS-CoV-2 several vibrant initiatives have been started as never before. For example, vaccine manufacturing front-runner come-up with mRNA vaccines [44], viral vector vaccine[45], classical attenuated vaccine etc. [46]. However, reports show COVID-19 vaccines are not going to be a silver bullet for the immunization of a community. And, for the long-run alternatives to the traditional therapeutics would be necessary as before [47, 48].
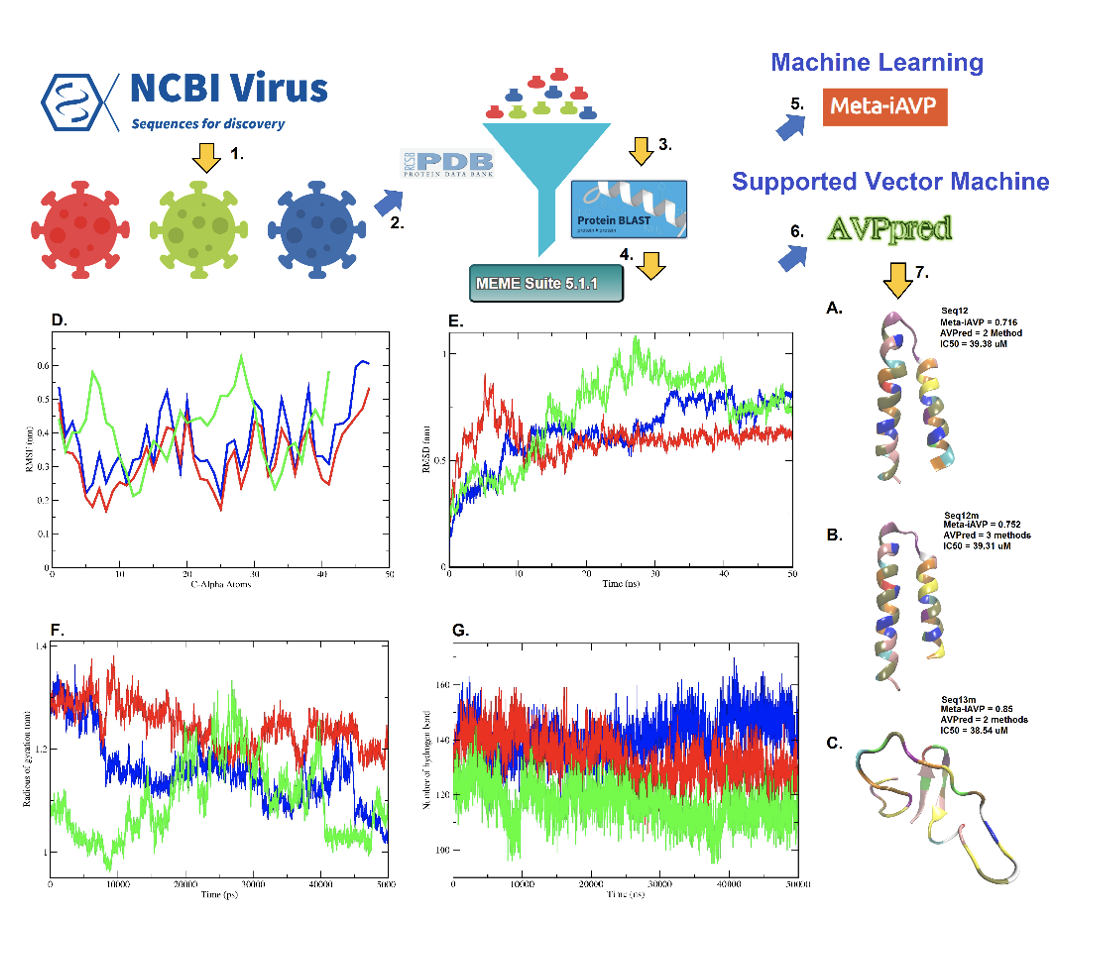
An antiviral peptide is one of such new alternatives [49]. And, antiviral peptides are successful to combat the SARS-CoV, and MERS-CoV [50, 51]. Therefore, with a similar kind of anticipation for the SARS-CoV-2, we have invented three new antiviral peptides against the SARS-CoV-2 (Figure 1a-c). Initially, the peptide sequences were interpreted as an antiviral peptide using AVPred [14], and Meta-iAVP [13]. The amino acid sequences of the predicted antiviral peptides were then mapped within the spike glycoprotein of the SARS-CoV-2. Figure 1d shows that antiviral peptides namely, Seq12, Seq12m, and Seq13m are in fact analogous peptides of the spike glycoprotein. Fasta sequences of the antiviral peptides are available in the supplementary section, Figure S1.
AVPred is an antiviral peptide prediction server that is based on a few sequence features viz., motifs, alignment, amino acid composition, and physicochemical properties [14]. Finally, the prediction of the antiviral peptide is made during 5-fold cross-validation using Supported Vector Machine (SVM). Antiviral peptides were predicted by the AVPred (based on the physicochemical model) can achieve up to 85% prediction accuracy with 0.70 Matthew’s Correlation Coefficient (MCC). However, the experimental validation dataset shows 86% prediction accuracy with 0.71 MCC.
Conversely, Meta-iAVP is based on a novel sequence-based meta-predictor with an effective features representation derived from Machine Learning Algorithms (MLA) and MLA types of features [13]. Interestingly, the use of MLA and MLA types features has increased the overall prediction accuracy, and MCC of 95.20%, and 0.90 respectively. At present more than 15 peptide-based drugs are in the pipeline of clinical trials [13].
The most worrying concern of an antiviral peptide is its immunogenic profile [52]. A low immunogenic profile is the desired characteristic of an antiviral peptide because a low immunogenic profile reduces the chances of elimination by the host defense system [53]. Therefore, we also investigate the immunogenic profile of the AVPs. Hopefully, none of the AVPs showed immunogenic properties to humans as a host. Furthermore, to intensify the immunogenic profile of the AVPs we have performed epitope prediction using IEDB tools. Soon after, epitope prediction, AVP sequences were mapped within the aligned sequence of the epitopes (only most frequently human alleles were chosen). Results showed that none of the epitopes have significant similarities with the AVPs (Figure S2). A few amino acid residues were apparently similar to the predicted epitopes. However, the binding postures (AVP-RBD) suggest these apparently similar amino acid residues were engaged with the interactions (AVP-RBD). Therefore, the probability of elimination by the host defense system is not considerable.
Furthermore, the AVPs in this present study are capable of inducing an anti-inflammatory cytokine, IL-4 [54]. But, they are not capable to induce IL-10 (a pro-inflammatory cytokine). In addition, AVPs were also non-allergenic as they do not have any known epitope for human IgE [20]. Other physicochemical properties viz., estimated half-life, instability index, water-solubility, theoretical IC50 values, etc. were also summarized in Table 1.
|
Table 1. Physicochemcial and biological properties of the antiviral peptides
|
|
Properties
|
Antiviral peptides
|
|
Seq12
|
Seq12m
|
Seq13m
|
|
APVpred1-5
|
AVP3,5
|
AVP3,4,5
|
AVP4,5
|
|
Meta-iAVP6
|
0.716
|
0.752
|
0.85
|
|
PROB-HemoPI7
|
0.17
|
0.17
|
0.46
|
|
Hydrophobicity
|
-0.23
|
-0.21
|
-0.10
|
|
Steric hindrance
|
0.69
|
0.67
|
0.65
|
|
Solvation
|
0.38
|
0.39
|
0.65
|
|
Hydropathicity
|
-0.78
|
-0.70
|
-0.25
|
|
Amphiphilicity
|
0.72
|
0.62
|
0.73
|
|
Hydrophilicity
|
0.09
|
0.03
|
-0.20
|
|
Net hydrogen
|
47.0
|
46.0
|
30.0
|
|
Charge (pH = 7)
|
1.1
|
0.0
|
-1.0
|
|
Isoelectric point (pI)
|
8.71
|
7.10
|
4.79
|
|
Molecular weight8
|
5302.68
|
5374.75
|
4657.83
|
|
Instability index9
|
37.74 s
|
46.15 u
|
53.32 u
|
|
Aliphatic index
|
72.77
|
72.77
|
76.10
|
|
GRAVY10
|
-0.777
|
-0.711
|
-0.0246
|
|
Estimated half life11
|
0.8 h
|
0.8 h
|
1 h
|
|
Water solubility
|
Good
|
Good
|
Poor
|
|
Epitope for IgE12
|
Non-allergen$
|
Non-allergen
|
Non-allergen
|
|
IL-4 inducer13
|
Non-inducer*
|
Non-inducer*
|
Non-Inducer*
|
|
IL-10 inducer14
|
Inducer$$
|
Inducer$$
|
Inducer$$
|
|
IC50
|
39.38 uM
|
39.31 uM
|
38.54 uM
|
|
1 Heamolytic prediction = PROB score HemoPI was calculated using HemoPI-1/1+motif, (SVM+Motif (HemoPI-2). Mapping of IgE epitopes and PID, MEME/MAST motif, Blast search on allergen representative peptides (ARPs). Half life = mamalian reticulocytes, in vitro. IL-4 inducer prediction are made using SMV method (SMV Thresold =0.5). *However, the complete antiviral sequence contain two motif to induce the IL-4 and hydride method (SMV+motif ) score is 1.04, suggesting it is IL-4 inducer peptide. Instability index9 : s = stable, u = unstable. GRAVY = Grand average of hydropathy.## The half maximal inhibitory concentration (IC50) = It a theoritical value calculated using IC50Pred. 11 Molecular weight in dalton. IL-10 inducing probability was calculated using RANDOM FOREST probability thresold = 0.5,and SMV method. AVPpred = , Meta-iAVP = . 2Motif, 3Align, 4Composition, 5Physicochemical composition
|
Recent work by Karoyan et al. [55] suggests hACE2 peptide mimics can block SARS-CoV-2 pulmonary cells infection with high affinity namely by P8, P9 and P10. In addition, we have found that the predicted IC50 [15] value of those high affinity peptides are 41.38 µM , 41.8 µM, and 42.06 µM respectively by P8, P9 and P10. (Supplementary file). However, anti-SARS-CoV-2 peptides viz., Seq12, Seq12m and Seq13m in this present study have predicted IC50 of only 39.38 µM, 39.31 µM, and 38.54 µM (Table 1) suggesting it might be promising too. Furthermore, other distinguished scientists have found anti-viral peptides from a variety of a different source such as, Fibronectin by Beddingfield et al. [56], Aprotinin by Bestle et al. [57], Glycopeptide antibiotic by Zhang et al. [58], mouse β-defensin-4 by Zhao et al. [59], PCSK target motif by Cheng et al.[60], HR2 by Xia et al. [61, 62], HR2 by Zhu et al.[63], heptad repeats 1 and 2 (HR1 and HR2) in the S protein by [64].
However, in this present study antiviral peptides were derived from the spike glycoprotein of the SARS-CoV-2. Besides, previous formal peer-reviewed works have failed to demonstrate how the studied peptides are effective after insertions of mutations in the RBD of the SARS-CoV-2. Here comes the uniqueness of the present study. In this present study molecular docking ( ), MM-PB/GBSA calculations ( ) suggest that the AVPs namely. Seq12, Seq12m, and Seq13m can sustain their properties with the mutant RBD model of the SARS-CoV-2. A recent study on the classification of the circulating strains of novel coronavirus 2019 suggests there are A, B, and C types of coronavirus. A-type coronavirus is ancient/old, B-type is a bit new meaning that it it has acquired dozens of new mutations and C-type is the current type, more contagious and has more mutations [65]. The results depicted from our present study suggest Seq12, Seq12m, and Seq13m might be effective to type A, B and C as well.
Antiviral peptides usually rupture the viral capsid, and eventually inhibit the viral replication cycles [66]. As mentioned earlier, the interactions between RBD-ACE2 is essential for viral entry inside the human host [67]. And recently, a research group led by Prof. Qiwei Zhang at the School of Public Health, Southern Medical University, China, has identified the molecular interactions between RBD-ACE2 [68]. Results suggest RBD includes six aromatic amino acid residues viz., TYR449, PHE456, PHE486, TYR489, TYR505, and TYR543. Five polar uncharged amino acid residues ASN487, GLN493, GLN498, THR500, and ASN501. And three non-polar aliphatic amino acid residues, LEU455, GLY496, and GLY502.
We utilized this information in molecular docking studies to zoom-in on the molecular interactions of the AVPs-RBD complexes. However, there is an increasing amount of fear of mutations in the RBD, that would probably make all anti-COVID-19 efforts nil [69]. Indeed, it is very true that if mutations incorporated into the RBD then therapeutics have to evolve accordingly. Therefore, we have constructed a few in silico mutant models of the receptor-binding domain (RBDm) of the SARS-CoV-2, such as energy mutant, evolutionary mutant, and combined mutant (Figure 2). The amino acid substitutions were incorporated based on Rosetta and FoldX [24]. And the results are summarized in Table 2. The combined mutant model suggested by the FireProt was further refined by the manual incorporations of amino acid residues based on the best energy substitutions suggested by FoldX, and Rosetta [24]. This combined approach has led to an increase in the free energy of the RBDm from - 27.63 kcal/mol (17 mutations) to -32.67 kcal/mol. And among the important amino acid residues of the RBD [67] only ASN487 is conserved (Figure S3).
|
Table 2. In silico mutant models of the receptor binding domain of the SARS-CoV-2
|
|
Combined mutant -27.63 kcal/mol (17 mutations)
|
Amino acid residues (Wild type to mutant type)
|
FoldX
(kcal/mol)
|
Rosetta
(kcal/mol)
|
|
A348P
|
-1.66
|
-2.34
|
|
N354E
|
-0.45
|
-
|
|
A372T
|
0.20
|
-
|
|
S373M
|
-2.30
|
-2.82
|
|
T393F
|
-1.41
|
-6.03
|
|
N394S
|
-0.63
|
-
|
|
S399M
|
-3.17
|
-3.09
|
|
R403K
|
0.48
|
-
|
|
K417V
|
0.21
|
-
|
|
N460K
|
-0.75
|
-
|
|
I468L
|
-0.40
|
-
|
|
T470Y
|
-2.10
|
-2.19
|
|
S477G
|
-0.03
|
-
|
|
S494Y
|
-1.47
|
-2.12
|
|
G502P
|
-1.94
|
-2.61
|
|
V503P
|
-1.18
|
-2.72
|
|
H519N
|
-0.02
|
-
|
|
Energy mutant: -25.41 kcal/mol (8 mutations)
|
A348P
|
-1.66
|
-2.34
|
|
S373M
|
-2.30
|
-2.82
|
|
T393F
|
-1.41
|
-6.03
|
|
S399M
|
-3.17
|
-3.09
|
|
T470Y
|
-2.10
|
-2.19
|
|
S494Y
|
-1.47
|
-2.12
|
|
G502P
|
-1.94
|
-2.61
|
|
V503P
|
-1.18
|
-2.72
|
|
Evolution mutant: -6.29 kcal/mol (12 mutations)
|
A348P
|
-1.66
|
-1.66
|
|
N354E
|
-0.45
|
-0.45
|
|
A372T
|
0.20
|
0.20
|
|
T393S
|
0.47
|
0.47
|
|
N394S
|
-0.63
|
-0.63
|
|
R403K
|
0.48
|
0.48
|
|
K417V
|
0.21
|
0.21
|
|
N460K
|
-0.75
|
-0.75
|
|
I468L
|
-0.40
|
-0.40
|
|
T470N
|
0.06
|
-0.06
|
|
S477G
|
-0.03
|
-0.03
|
|
H519N
|
-0.02
|
-0.02
|
The structural analysis of the receptor-binding domains (RBD) of the SARS-CoV-2 has been summarized in Figure 2. The RRDis Map analysis shows that RBDm and RBD can be overlapped. Moreover, the RRDis Map showed that RBD and RBDm had an average distance of 51.74 (SD:1.486, a secreted site of the map), suggesting they are structurally different (Figure S5). Moreover, the RBDb/c/e structurally distinguished from RBDm/RBD and they can not be overlapped with either RBD or RBDm. The RRDis map showed that the average distance of the RBDb/c/e was 28.884 (SD:0.032, a secreted site of the map), suggesting they are structurally close to each other. In addition, the correspondence analysis also suggests RBDb/c/e are structural neighbors and, RBD and RBDm are structurally distinguished from RBDb/c/e. The structural tree (Supplementary Figure S4) showed that RBDb, RBDc, and RBDe are orthologous. Conversely, RBD/RBDm are paralogous. Moreover, a comparison of all four mutant types of the RBD by selecting the wild type RBD as a reference, revealed that all models have exactly the same RMSD value of 1.7 except RBDe RMSD = 1.5). Besides, RBDc was 96% identical with the wild type RBD followed by RBDm (94%), RBDe (88%), and RBDb (84%) and the Dali Z-scores were 25.3, 25.3, 25.2, and 23.9 respectively for RBDm, RBDc, and RBDe, and RBDb (Table 3).
|
Table 3. Structural analysis of different mutant model of the receptor binding domain (RBD) of the SARS-CoV-2
|
|
Sl. No.
|
Mutant models of RBD
|
RMSDa
|
Percentage of identitiesb
|
Z-score*
|
|
1.
|
RBDm
|
1.7
|
94
|
25.3
|
|
2.
|
RBDc
|
1.7
|
96
|
25.3
|
|
3.
|
RBDe
|
1.5
|
88
|
25.2
|
|
4.
|
RBDb
|
1.7
|
84
|
23.9
|
|
aRMSD = root mean square deviations, bPercentage of identities with the wild type RBD structure, RBDm = mannual mutant, RBDc = combind model, RBDe = energy mutant, RBDb = evolutionary mutant model of the receptor binding domain. *Dali Z-score.
|
Results from molecular docking studies showed that AVPs-RBD had a thermodynamically favorable interaction (Table 4). Moreover, the AVPs were engaged with nearly all-important amino acid residues of the RDB as mentioned in the earlier report [67] . Out of three antiviral peptides of this present study, Seq12 (47 mer) showed an astonishing HADDOCK score of -111.2 kcal/mol followed by Seq13m (81.4 kcal/mol), and Seq12m (76.8 kcal/mol) suggesting AVPs are well-docked with the RBD (Figure 3, S6). In addition, molecular docking studies between RBDm and the antiviral peptides (RBDm-AVP) summarized in Table 4. Results suggest Seq12, Seq12m, and Seq13m can retain the thermodynamically favorable binding properties with the mutant RBD. Moreover, Seq12 surprisingly showed an improved binding free energy (kcal/mol) in comparison to the wild type RBD (kcal/mol). Further, Seq12, and Seq12m have negligible cytotoxicity (0.17; while 1.0 or higher is considered as toxic and can rapture the human RBCs), (Table 1). These findings combinedly justify the novelty of the AVPs, and its possible therapeutic use in the future.
|
Table 4. HADDOCK docking properties of the antiviral peptides complex with the receptor binding domain of the SARS-Cov-2
|
|
Properties
|
Systems
|
|
Seq12 + RBD
|
Seq12 + RBDm3
|
Seq12m + RBD
|
Seq12m + RBDm
|
Seq13m + RBD
|
Seq13m + RBDm
|
|
HADDOCK Score
|
-111.2
|
-92.8
|
-76.8
|
-76.7
|
-81.4
|
-41.7
|
|
Cluster size
|
104
|
36
|
13
|
27
|
40
|
14
|
|
RMSD1
|
0. 5
|
0.4
|
2.6
|
4.7
|
0.5
|
3.5
|
|
VDW2 energy
|
-68.8
|
-71.3
|
-71.9
|
-78.3
|
-75.2
|
-63.4
|
|
Electrostatic energy
|
-182.4
|
-142.0
|
-145.0
|
-53.2
|
-70.3
|
-73.8
|
|
Desolvation energy
|
-36.9
|
-36.6
|
-4.9
|
-43.1
|
-20.9
|
-28.5
|
|
1RMSD = Root mean square deviation from the overall lowest-energy structure. 2VDW = Van der Waals energy. 3RBDm = mutant model of the receptor binding domain of the SARS-CoV-2. Energy units are in kcal/mol.
|
SARS-CoV-2 has a structural membrane protein called Protein-M [70]. Protein-M is a tri-pass transmembrane protein (Figure 4) which is essential for re-assembling viral structural units into a mature virus [71]. Therefore, Protein-M also embraces importance as a potential therapeutic target. Results from our investigation indicate AVPs are also participating in a thermodynamical favorable interactions with the TM-1 region followed by TM-2 and TM-3 respectively (Figure 4a). In addition, the molecular docking studies between the AVPs and the active site of the viral RNA-dependent RNA polymerase (RdRp) were not thermodynamically favorable interaction (Supplementary section). We speculate that the antiviral peptide Seq12, Seq12m, and Seq13m acts as an anti-SARS-CoV-2 peptide by two possible mechanisms. Firstly, by inhibiting the RBD-ACE2 interaction. And secondly, by binding with M-protein followed by an eventual inhibition of the viral re-assembly/re-packaging.
Results, derived from MD-simulation of the studies of the AVPs are summarised in Figure 5. The root mean square deviation (RMSD) of alpha carbon atoms of all systems are analyzed to detect their stabilities. It is observed from Figure 6 that Seq12m, has the lowest RMSD value than Seq12 and Seq13m respectively. Antiviral peptides namely Seq12, Se12m, and Seq13m have ≈ 0.274 nm, ≈ 0.274 nm, and ≈ 0.286 nm of RMSD values at 1 ns. The highest RMSD values are respectively ≈ 0.852 nm, ≈ 0.908 nm, and ≈ 1.074 nm for Seq12, Seq12m, and Seq13m. A closer look at the RMSD plot suggests Seq12m has the highest fluctuation from 5 ns - 10 ns and gets stabilized throughout the trajectory. However, at the start, RMSD-fluctuation is lower in the case of Seq12 and the highest RMSD-fluctuation is observed between 30 ns - 40 ns. Quite similarly, the RMSD-fluctuation of Seq13m is highest between 20 ns -40 ns. Root means square fluctuation (RMSF) helps to understand the flexibility of each amino acid residue [72]. Seq13m is found to have the highest RMSF from the 5th to 7th and 25th to 30th position of the amino acid sequence.
The lower degree of fluctuation with its consistency through the simulation indicates the greater compactness and rigidity of a system [72]. Rg of Seq13m is fluctuated from 10 ns -40 ns and reaches the highest value of 1.331 at ≈ 25 ns. However, Rg values of Seq12 and Seq12m are very similar at the beginning and remain same up to 10 ns. Further, Rg fluctuations are prominent after 10 ns and sustain such dissimilarities throughout the trajectory. The lowest Rg values are ≈ 1.041 nm (50 ns), ≈ 1.380 nm ( before 10 ns), and ≈ 0.944 nm ( before 10 ns) respectively for Seq12, Seq12m, and Seq13m. In summary systems i.e., Seq12 and Seq12m showed better stability throughout the complete trajectory. The number of inter water-peptide hydrogen bonds in the simulated systems were also compared. The highest number of hydrogen bonds are formed respectively by Seq12 (40 - 45 ns), Seq12m (15 ns), and Seq13m (before 5 ns).
In addition, MD-simulation studies of the AVP-RBD complex systems (antiviral peptide + RBD/RBDm) of the 100 ns MD production for piror to the MM-PBSA analyses were summarised in Figure 6. RMSD, and RMSF were stable throughout all MD-trajectory and were most stable in the case of Seq12-RBD/RBDm, and Seq12m-RBD/RBDm complex. However, RMSD values fluctuates in the case of Seq13m-RBD/RBDm complex but the these values were within acceptable range of 3nm suggesting extension of the simulation time scale is not required. RMSF value is a measures of the avarage deviations particular atoms or group of atoms from the initial reference structure [73]. Results showed that, in case of the Seq13m+RBDm, RMSF fluctuation was high from about 138th -158th position and last twenty five residues of the RBD. Similarly, in the case of Seq13m+RBD the RMSF value highly fluctuates from about 200th to rest of the residues of the RBD. However, only two important interacting amino acid residues namely, PHE486 and TYR489 was remain within the flutuating regions (Figure 6b). Besides, the ΔGbind of Seq13m+RBDm and Seq13+RBD were also very good in comparison to the other RBD-AVP complex systmes (Table 5) suggesting this little stuctural deviations does not have negative impacts on binding. The radious of gyrations (Rg) around the axis were stable (1.16-1.47 nm) for all complex systems, Besides, Rg of the AVP-RBD complexes were overlapping in the case of all complex systems except Seq12m-RBD. In addition, the solvent accessible surface area (SASA) of the Seq13m-RBD/RBDm complex systems were highest (16425.50 nm2), and this value was lowest (13454.30 nm2) for the Seq12m-RBDm complex system. Moreover, the number of hydrogen-bonds formed during the MD-simulation run was limited in the case of inter AVP-RBD/RBDm complex (0-10). However, during the entire MD-simulation run more H-bonds (0-99) were formed between protein complex and water.
The results obtained from MM-PB/GBSA analysis are also in agreement with the molecular docking studies (Table 5). Moreover, the binding free energies (MM-GBSA) per amino acid residues (only the top twenty participating amino acid residues) are summarized in Figure 7. It shows that TYR489 was an important amino acid residue of the RBD because TYR489 was a common contributor to the lowest binding free energies of Seq12-RBD (-9.30 kcal/mol), Seq12m-RBD (-6.55kcal/mol), and Seq13m-RBD (-4.91kcal/mol). On the other hand, GLU24 (-9.20 kcal/mol) of Seq12, THR20 (-3.36kcal/mol) of Seq12m, and PRO25 (-6.56 kcal/mol) of Seq13m were critically important for the respective antiviral peptides for the same reasons. Further, GLN498 (-0.22 kcal/mol), and GLN493 (-0.28 kcal/mol) were poor contributors for the antiviral peptide-RBD (AVP-RBD) interactions respectively for Seq12 and Seq12m. However, GLY496 of the RBD poorly contributes to the Seq13m-RBD interaction.
Conversely, Seq12-RBDm, Seq12m-RBDm, and Seq13m-RBDm interactions reveal a different picture (Figure 7). For example, amino acid residues of mutant RBD namely, TYR501 (-6.32 kcal/mol), MET494 (-5.11 kcal/mol), and TYR-450 (-5.24 kcal/mol) contributes lowest binding free energy changes while interacting with Seq12, Seq12m, and Seq13m respectively. It is indeed, noticeable that in all cases (wild and mutant RBD) tyrosine contributes the lowest binding free energy irrespective of its positions in the RBD. The biochemical and biophysical properties of tyrosine residue enable itself for this phenomenon. However, tyrosine has not participated in the top twenty amino acid residues in the case of Seq12m-RBDm. On the contrary, LEU493 was a poor contributor to the binding free energies of Seq12-/ Seq12m-RBDm interactions. However, ASN449 was another poor contributor in the case of Seq13m-RBDm interaction. Overall, the molecular details of AVP-RBD/RBDm interactions calculated using MM-GBSA provide evidence for the fact that AVPs were occupied the important amino acid residues of the RBD which are critical for the RBD-ACE2 interactions [67].
In addition, the MM-PBSA analyses suggest Seq12m-RBDm has the best ΔGMMPBSA (-84.75 kcal/mol), followed by Seq13m-RBDm (-79.30 kcal/mol), and Seq12m-RBDm (-78.72 kcal/mol), etc. (Table 5). Moreover, the linear regression fit between predicted ΔGPB/GBSA calculated for 50 ns and predicted ΔGPB/GBSA calculated for 100 ns is summarized in figure 8. It showed that the R2 of the regression model of the ΔGGBSA was 0.97 (p = 0.0003) suggesting ΔGbind are significantly correlated with the model. However, in the case of ΔGPBSA the R2 was 0.81 (p = 0.0149), although all variables were within 95% confidence intervals. This is because we have found that the ΔGPBSA calculated for 50ns and 100ns have the highest difference in the case of Seq13m-RBD (ΔGPBSA calculated for 50 ns was -29.22 kcal/mol and ΔGPBSA calculated for 100 ns was -42.52 kcal/mol), (Table S1).
|
Table 5. Binding free energy (ΔGbind) of different AVP-RBD complex systems calculated using MM-GB/PBSA
|
|
Systems
|
MM-GBSA*
|
MM-GBSA**
|
MM-PBSA#
|
|
Seq12 + RBD
|
-92.09
|
-45.37
|
-49.80
|
|
Seq12 + RBDm$
|
-93.31
|
-53.64
|
-84.75
|
|
Sqe12m + RBD
|
-50.09
|
-61.07
|
-61.84
|
|
Sqe12m + RBDm$
|
-75.13
|
-62.96
|
-78.72
|
|
Seq13m + RBD
|
-57.96
|
-40.71
|
-42.52
|
|
Seq13m + RBDm$
|
-53.90
|
-38.59
|
-79.30
|
|
* MM-GBSA was calculated using HawkDock.
# MM-PBSA was calculated using CaFE tools with the MD-trajectory generated using NAMD software.
** MM-GBSA was calculated using Abmer 16 package Generalized Born ESURF calculated using 'LCPO' surface areas.
$ Combined mutant model of receptor binding domain is manually optimized to best energy model (RBDm).
Energy units are in kcal/mol.
|
{kind=link}