Reagents
2-DG (FUJIFILM WAKO, Tokyo, Japan), CQ (Sigma-Aldrich, St. Louis, MO, USA), and iadademstat (ORY-1001) 2HCL (Selleckchem, Houston, TX, USA) were dissolved in milliQ water. SP-2509 (Merck, Darmstadt, Germany), OG-L002 (Selleckchem), T-3775440 HCL (Selleckchem), DCA (Tokyo Kasei Corp., Tokyo, Japan), perhexiline maleate salt (Cayman Chemical Company, Ann Arbor, MI, USA), and oligomycin (Sigma-Aldrich) were dissolved in dimethyl sulfoxide (DMSO; Nacalai Tesque, Kyoto, Japan).
Cell culture
PANC-1, PK-1, and KLM-1 cells were purchased from the RIKEN Cell Engineering Division (RIKEN BioResource Research Center, Tsukuba, Japan). Cells were maintained in RPMI-1640 (Nacalai Tesque, Inc., Kyoto, Japan) supplemented with 10% fetal bovine serum (Life Technologies, Grand Island, NY, USA) and antibiotics (Nacalai Tesque Inc., Kyoto, Japan). KPC cell line (C57/BL6 genetic background) was purchased from CancerTools (London, UK). Cells were maintained in DMEM (Nacalai Tesque) supplemented with 10% fetal bovine serum (Life Technologies, Carlsbad, CA, USA) and antibiotics (Nacalai Tesque). Cells were cultured at 37°C in a humidified chamber with 5% CO2. PANC-1 cells were cultured in medium supplemented with glucose (2 g/L), galactose (2 g/L), or low levels of glucose (0.2 g/L). PK-1 and KLM-1 cells were cultured in medium supplemented with glucose (2 g/L) or low levels of glucose (0.2 g/L). KPC cells were only cultured in 4.5 g/L glucose.
Metabolic compound screening
An Anti-Cancer Metabolism Compound library (96-well, catalog number L2130) was purchased from TargetMol (Wellesley Hills, MA, USA). PANC-1 cells were cultured in glucose (2 g/L), galactose (2 g/L), or low-glucose (0.2 g/L) medium with 130 different types of metabolic compounds for 48 h. To assess cell viability, the cells were then incubated with 0.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Dojindo Laboratory, Kumamoto, Japan) for 2 h. After incubation, the formazan crystals produced were dissolved in 200 µL DMSO. The absorbance of the resulting solution was measured at 540 nm to determine the amount of MTT-formazan formed.
Measurement of intracellular ATP concentration
Intracellular concentration of ATP was assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer's protocol.
Western blotting
Cells were lysed on ice in lysis buffer [PBS (pH 7.4) containing 1% Triton X-100 (Nacalai Tesque) and a protease inhibitor cocktail (Roche, Mannheim, Germany)]. Equal amounts of protein from each sample were separated by SDS-PAGE and transferred to a polyvinylidene difluoride membrane (Millipore, Berlin, Germany). The membranes were blocked with 5% bovine albumin (Nacalai Tesque) or 5% nonfat dry milk (Cell Signaling Technology, Beverly, MA, USA) at 25 ℃ for 1 h and probed overnight with primary antibodies (Cell Signaling Technology) specific for H3K4me3, LC3B (D11), LSD1 (C69G12), phospho-PDH α1 (Ser293), PDH, or β-actin. Immunolabeled proteins were detected using horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Buckinghamshire, UK) and ECL Prime detection reagents (GE Healthcare). The signals were visualized using an ImageQuant LAS 4000 (Version 1.3) system (GE Healthcare).
Transfection of siRNA
siRNA oligonucleotides (Japan Bio Services Co. Ltd., Saitama, Japan) were transfected at final concentrations of 100 nM using Lipofectamine 2000 (Invitrogen, Waltham, MA, USA). Details of oligonucleotide sequences are presented in Supplementary Table 2.
RNA isolation and quantitative real-time PCR
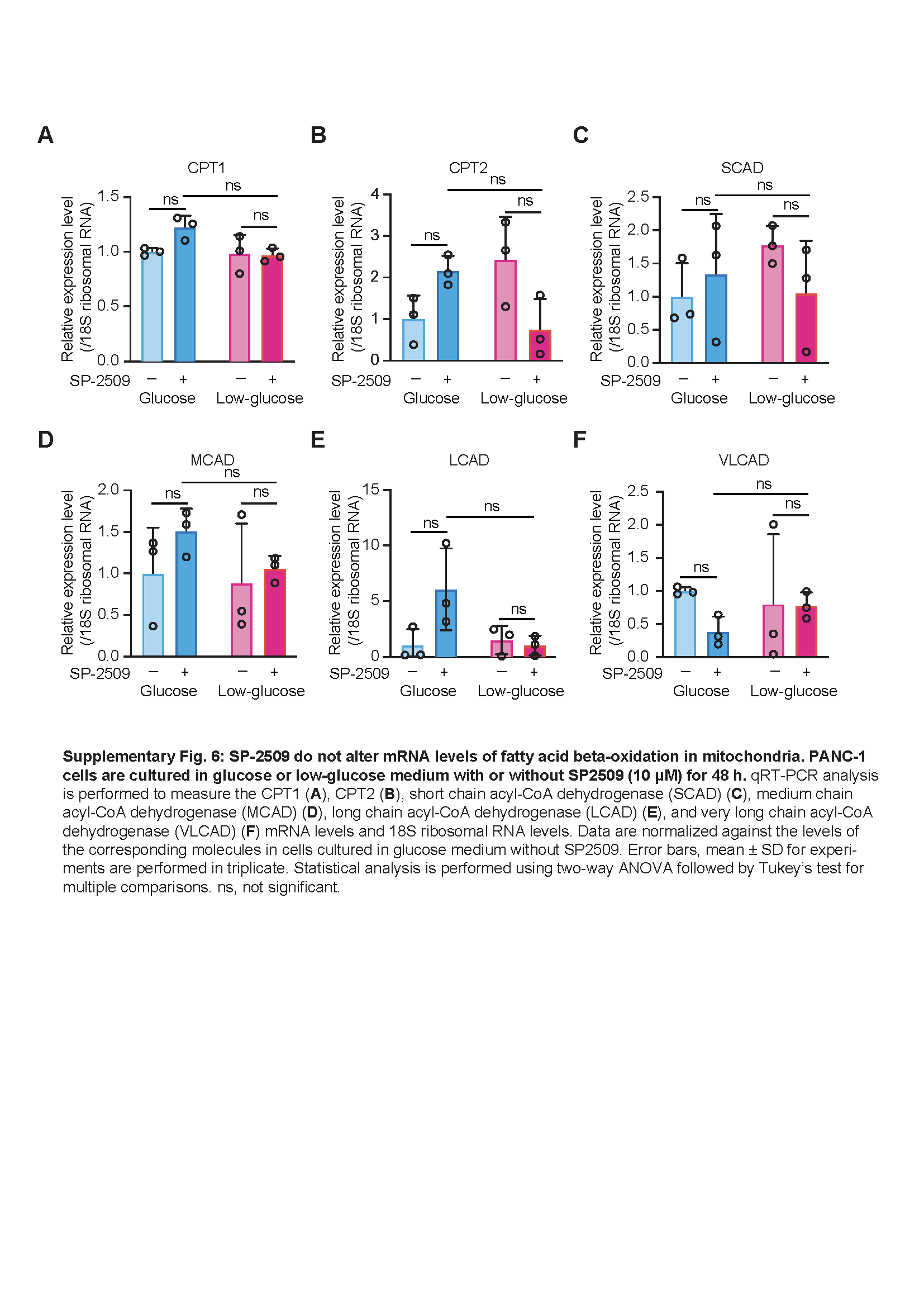
Total RNA was isolated from PANC-1 cells using Sepasol-RNA I reagent (Nacalai Tesque) and reverse-transcribed using ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan). The resulting cDNA was mixed with THUNDERBIRD quantitative real-time PCR mix (TOYOBO). The mixture was then subjected to quantitative real-time PCR. Primers used in this study are listed in Supplementary Table 3. The cycling conditions were as follows: 95°C for 60 s, followed by 40 cycles (50 cycles for long-chain acyl-CoA dehydrogenase) of 95°C for 10 s and 60°C for 60 s. Relative expression of mRNA was calculated after normalization against the levels of 18S ribosomal RNA or β-actin.
Measurement of cellular lactate
Cellular lactate levels were measured using a Lactate Assay Kit-WST (Dojindo Laboratory).
Proteomic analyses based on nano liquid chromatography-tandem mass spectrometry (NanoLC-MS/MS) and bioinformatics analysis
PANC-1 cells were cultured in glucose or low-glucose medium for 6 days. Cells were lysed with ice-cold lysis buffer [PBS (pH 7.4) containing 1% Triton X-100, protease inhibitor, and phosphatase inhibitor cocktail (Roche)]. Proteins were precipitated from cell lysates using the ProteoExtract Protein Precipitation Kit (Millipore), according to the manufacturer’s guidelines. Precipitated proteins were dissolved in approximately 10 µL of 50 mM ammonium bicarbonate buffer (pH 8.1) containing 0.1% RapiGest SF (Waters Corporation, Milford, MA, USA). Total protein concentrations were determined using Pierce™ 660 nm protein assay reagent (Thermo Fisher Scientific), including Ionic Detergent Compatibility Reagent (Thermo Fisher Scientific, Waltham, MA, USA). To each 0.5 mL tube, 10 µg of protein was aliquoted and diluted with 25.5 µL of 50 mM ammonium bicarbonate buffer (pH 8.1) containing 0.1% RapiGest SF and 1.5 µL of dithiothreitol (100 mM in distilled water; Nacalai Tesque). The solution was heated at 60°C for 30 min. After cooling to 25°C, 3 µL of iodoacetamide (100 mM in distilled water; FUJIFILM WAKO) was added to the solution (final concentration was 10 mM), and the tubes were incubated at room temperature for 30 min in the dark. Next, the samples were digested with trypsin (mass spectrometry-grade, Promega; protein/enzyme = 20/1, w/w) at 37°C overnight. The solution was quenched with 10% TFA (pH < 3) and incubated at 37°C for 30 min. After centrifugation (13 300 × g, 10 min, 25°C), the solution was desalted using GL-Tip SDB (GL Science, Tokyo, Japan) according to the manufacturer’s instructions. The eluate was dried in vacuo and dissolved in distilled water containing 2% MeCN and 0.1% formic acid.
For proteomic analysis, nanoLC-MS/MS analyses were performed on an Ultimate 3000 RSLCnano system (Thermo Fisher Scientific) coupled to a Q Exactive hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific) equipped with a nano-electrospray ionization source. The nanoLC system was equipped with a trap column (Thermo Fisher Scientific) and analytical column (Nikkyo Technos, Tokyo, Japan). Peptide separation was performed using a 90 min gradient between water containing 0.1% formic acid (mobile phase A) and acetonitrile containing 0.1% formic acid (mobile phase B) at a flow rate of 300 nL/min. The elution was set as follows: 0–3 min, 2% B; 3–63 min, 2–40% B; 93–95 min, 40–95% B; 95–105 min, 95% B; 105–107 min, 95–2% B; 107–120 min, 2% B. The mass spectrometer was operated in data-dependent acquisition mode. MS parameters were set as follows: spray voltage, 2.0 kV; capillary temperature, 275°C; S-lens RF level, 50; scan type, full MS; scan range, m/z 350–1500; resolution, 70 000; polarity, positive; automatic gain control target, 3 × 106; and maximum injection time, 100 ms. MS/MS parameters were set as follows: resolution, 17 500; automatic gain control target, 1 × 105; maximum injection time, 60 ms; normalized collision energy, 27; dynamic exclusion, 15 s; loop count, 10; isolation window, 1.6 m/z; and charge exclusion, unassigned, 1, 8, > 8. Measurements were performed in duplicate for each sample and all replicated data were merged and used for quantitative analysis.
Protein identification and relative quantitation were performed using the Proteome Discoverer 2.4 SP1 (Thermo Fisher Scientific). Search parameters were as follows: search engine, Sequest HT; protein database, SwissProt (Homo sapiens); enzyme name, trypsin (full); dynamic modification, oxidation (methionine, + 15.99 Da); static modification, carbamidomethyl (cysteine, + 57.02 Da); precursor mass tolerance of 10 ppm; and fragment mass tolerance of 0.02 Da. The label-free quantification parameters for the detected peptides were set as follows: precursor quantification, precursor abundance based on area, normalization mode, and total peptide amount. Gene symbols of 199 proteins upregulated under low-glucose conditions were used for pathway enrichment analysis using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost). The raw data are presented in Supplementary Table 4. Ranking top eight pathways from KEGG and WikiPathways were displayed in the main figure. The abundance of proteins under glucose or low-glucose conditions was used for analysis with GSEA 4.3.2, using the following settings: number of permutations = 1000, permutation type = gene set, enrichment statistics = weighted, metric for ranking genes = ratios of classes.
Immunofluorescence
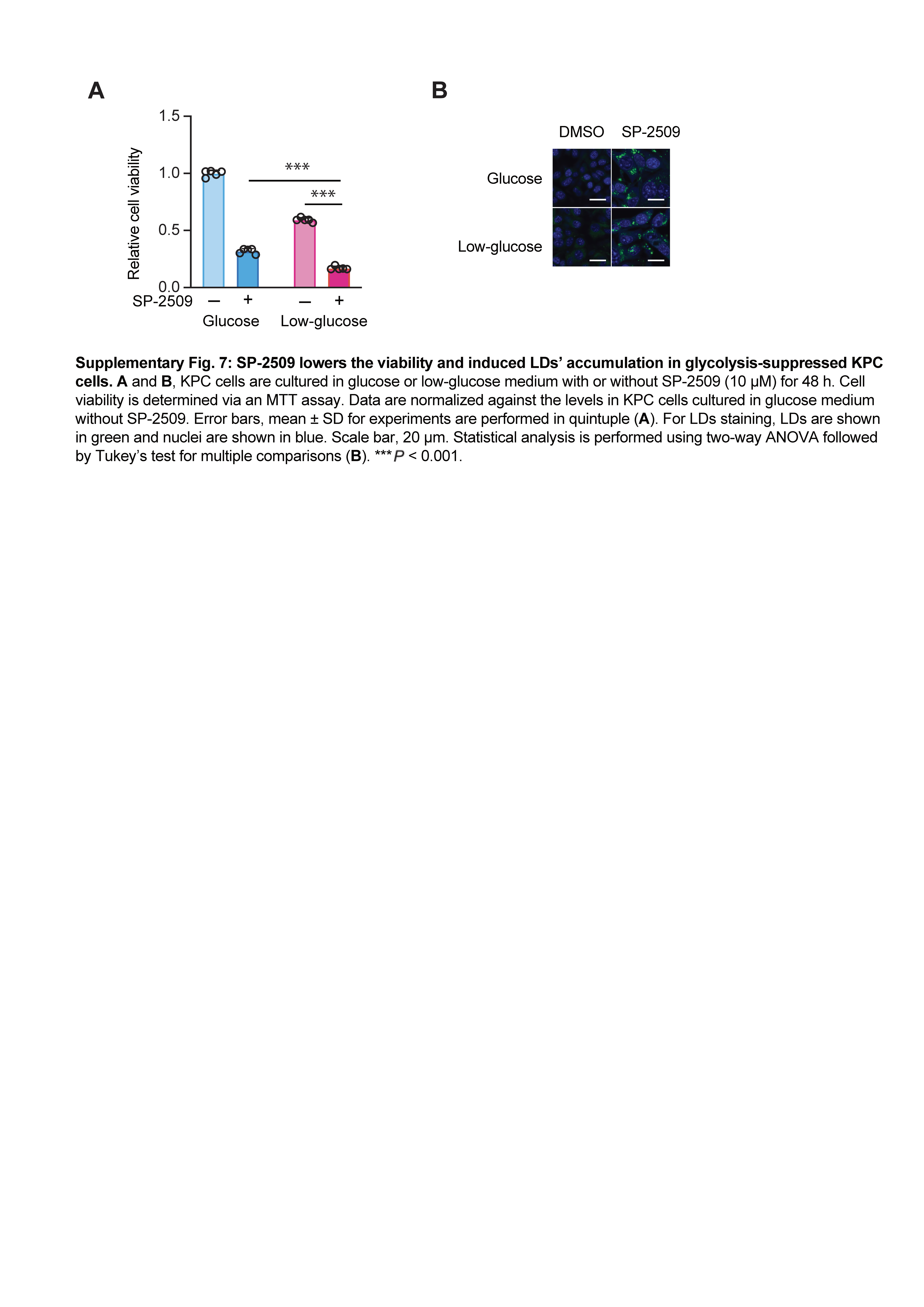
The cells were seeded in 35 mm glass bottom dishes (MATSUNAMI, Japan) and left overnight to attach. After CQ or SP-2509 treatment, cells were incubated with LysoTracker (1 µM) (Thermo Fisher Scientific, Hanover Park, IL, USA) for 1 hour. After that, cells were fixed in 4% paraformaldehyde for 10 min. Fixed cells were stained with 1 µg/mL BODIPY-493-/503 (Thermo Fisher Scientific) and 5 µg/mL Hoechst 33342 for 30 min at 37 ℃. All samples were imaged using a Carl Zeiss LSM780 laser scanning confocal microscope (Prenzlauer, Berlin, Germany). Images of five random fields of vision from each group were captured and analyzed using Image J. Ratio of average areas of LDs/cell number was used to indicate the level of LDs.
Immunohistochemical
For mouse samples, Immunofluorescence staining of α-SMA, CD4 and CD8 was done as previously described [43]. Rabbit anti-α-SMA (Abcam Cambridge, U.K.; dilution of 1:200), rabbit anti-CD4 (Abcam; dilution of 1:500), rat anti-CD8 (Abcam; dilution of 1:200), Alexa Fluor 488-conjugated secondary Ab (Abcam; dilution of 1:200), Alexa Fluor 546 conjugated secondary Ab (Thermo Fisher Scientific; dilution of 1:200). All samples were imaged using a Carl Zeiss LSM780 laser scanning confocal microscope (Prenzlauer, Berlin, Germany). Images of five random fields of vision from each group were captured and analyzed by Image J.
Animal Treatment Protocols
Female CB.17SCID mice (12 pups) and C57BL/6 mice (36 pups) aged 6 weeks were purchased from The Jackson Laboratory (Kanagawa, Japan) and maintained in an experimental animal facility at Chiba University (Chiba, Japan). For CB.17SCID mice, each mouse was subcutaneously administered with 1.5×106 PANC-1 cells suspended in 50 µL serum-free RPMI-1640 and 50 µL Cultrex™ Basement Membrane Extract, Type3, PathClear™ (bio-techne, Minneapolis, MN, USA). After 7 days, the tumor volume was over 100 mm3 and the mice were randomly divided into the following four treatment groups: untreated control, 2-DG alone, SP-2509 alone, and a combination of 2-DG and SP-2509. SP-2509 (0.5 mg/mouse) was administered intraperitoneally to the mice twice per week, and 2-DG (10 mg/mouse) was administered intraperitoneally three times per week. Tumor volumes were calculated using the following standard formula: width2×length×0.52 [44], and were measured every 2–3 days using Mitutoyo digital calipers (Mitutoyo, Tokyo, Japan). The tumor sizes were controlled so that their volumes did not exceed 2000 mm3. For C57BL/6 mice, each mouse had their pancreas orthotopically injected with 5 ×105 KPC cells suspended in 50 µL HBSS buffer. After 7 days, the mice were randomly divided into the following four treatment groups: untreated control, 2-DG alone, SP-2509 alone, and a combination of 2-DG and SP-2509. SP-2509 (0.5 mg/mouse) was administered intraperitoneally to the mice twice per week, and 2-DG (10 mg/mouse) was administered intraperitoneally three times per week.
Oil Red O staining
Oil Red O staining was conducted as previously described [45]. Samples were imaged using ECLIPSE Ci-L plus Upright Microscope (NIKON, Tokyo, Japan).
Statistical analysis
All data are presented as mean ± SD of at least three independent experiments, unless indicated otherwise. Statistical analysis was performed using an unpaired Student's t-test, Log-rank (Mantel-Cox) test or two-way ANOVA followed by Tukey’s test. P value < 0.05 was considered significant. All analysis were conducted using GraphPad Prism version 9 (Dotmatics, Boston, MS, USA)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}