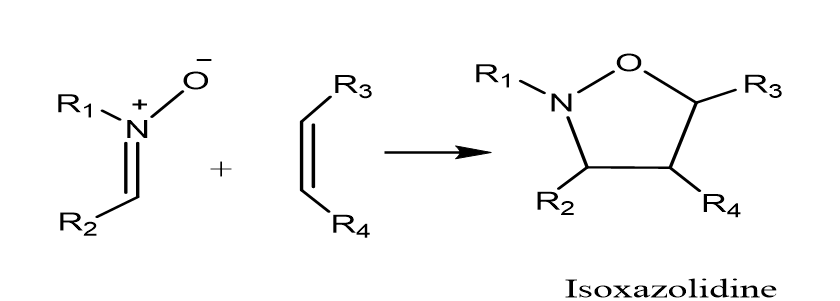
In our research, our main objective is to offer a theoretical basis for the observed regioselectivity and stereoselectivity of the nitrone (a1) and alkene (b1) reaction depicted in Scheme 2. To accomplish this goal, we utilize various theoretical approaches, which include determining activation energies, employing the frontier molecular orbital (FMO) theory, and using reactivity indices derived from conceptual density functional theory (DFT). It is essential to emphasize that the reaction being studied is kinetically controlled, indicating that the final outcome of the reaction is governed by the relative activation energies of the different competing reaction pathways.
3.1. Prediction of NED/IED character
To clarify the NED (Normal Electron Demand) or RED (Reverse Electron Demand) characters of the cycloaddition reactions between the nitrone and a olefin, we calculated: the HOMO/LUMO gap energies for the two possible combinations between the dipole and the dipolarophile, the chemical electron potential µ, the electrophilicity index ω, and the nucleophilicity index (N) (see table.2). Table 2 shows that the gaps of the energies corresponding to the HOMO (dipole)/LUMO (dipolarophile) combination (7.30 eV) between dipole (a1) and the dipolarophile (b1) is greater than this corresponding to the HOMO (dipolarophile)/LUMO (dipole) combination (5.08 eV) (see Fig. 1). In other hand, the alkene has the lowest electrophilicity index (ω = 0.57) and the highest electronic potential (µ = -0.110) compared to the dipole (ω = 1.66, µ = -0.148). These results indicat that the dipolarophile (alkene) behave as an electron donor and dipole (nitrone) as an electron acceptor. Therefore, the 13DC reaction carry a RED (reverse electron demand) character. It can be seen that the alkene act as a nucleophile while nitrone acts as an electrophile. Therefore, the charge transfer on this reaction will take place from the alkene to the nitrone, in complete agreement with the calculated charge transfer analysis at TSs (see later).
Table 1
Border orbital energies ( HOMO /LUMO) and global properties of dipole (a1) and dipolarophile (b1) .
| |
HOMO
|
LUMO
|
µ
|
η
|
ω
|
DNmax
|
N
|
|
a1
|
-0,2378
|
-0,0581
|
-0,1480
|
0,1797
|
1,66
|
0,824
|
2,65
|
|
b1
|
-0,2452
|
0,0307
|
-0,1073
|
0,2759
|
0,57
|
0,389
|
2,45
|
3.2. Prediction and rationalization of experimental regioselectivity
In order to predict and rationalize the experimentally observed regioselectivity of the 13DC reaction between nitrone 1 and alkene 2 we used two theoretical approaches :
a- Activation energy calculations
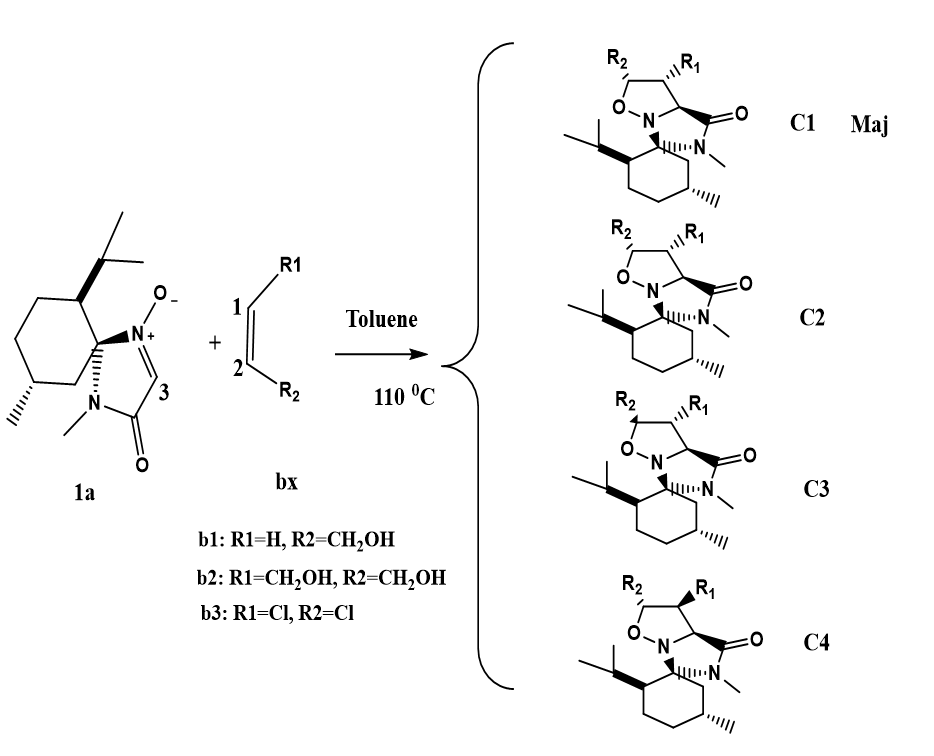
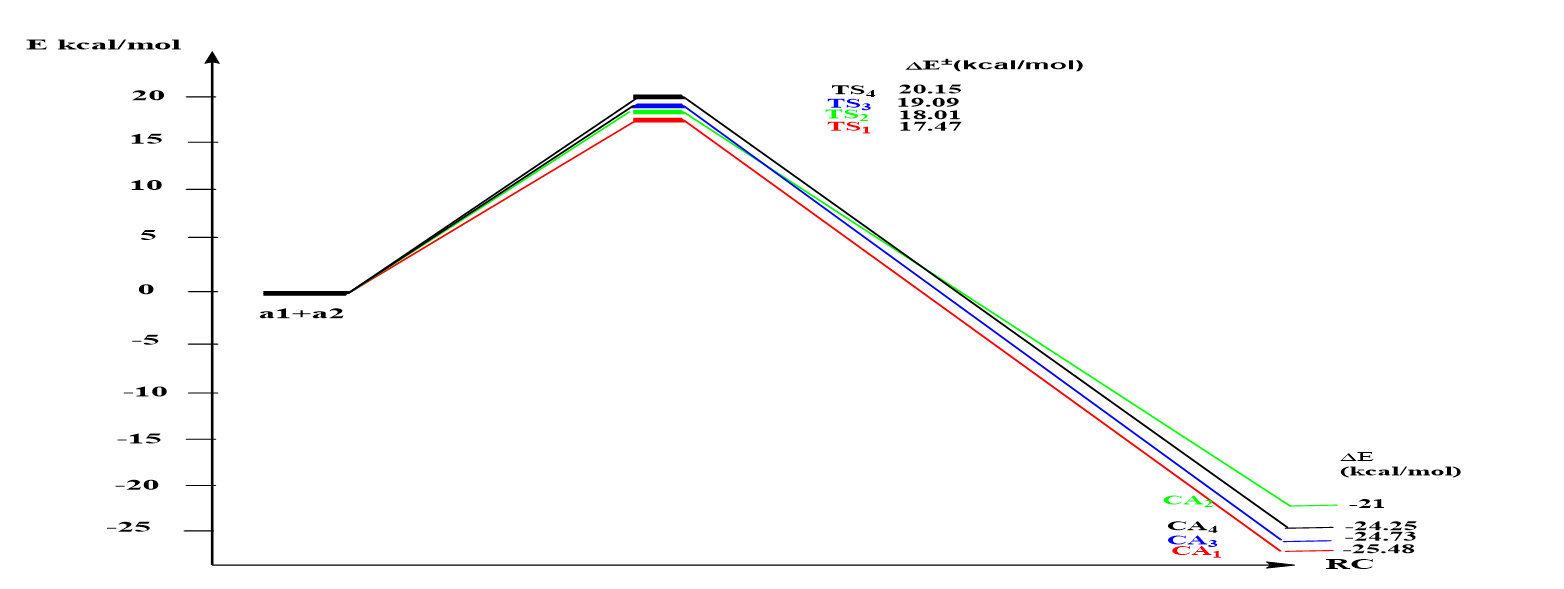
The interaction between nitrone 1a and alkene 2a at the 13DC reaction can result in four isomeric structures, as depicted in Scheme 2. To investigate the 13DC reaction, Density Functional Theory (DFT) is employed. We present a computational analysis of the regioselectivity and stereoselectivity of the nitrone 1a (dipole) cycloaddition to alkene 2a (dipolarophile). Our primary objective is to calculate the energy barrier for the 13DC reaction. In these computations, the B3LYP/6-31G* method is utilized with toluene as the solvent. Figure 2 displays the transition states corresponding to the two cyclization modes, 1.3 and 2.3. The activation energy of TS1 (17.46 kcal/mol) is lower than that of TS2 (18.01 kcal/mol) when the spatial position of the R2 = CH2OH group is in backwar. Similarly, the activation energie for TS3 (19.09 kcal/mol) is lower than that of TS4 (20.15 kcal/mol) when the spatial position of the group R2 = CH2OH is in the forward (see Scheme 3). These results demonstrate that the regioisomers 1.3 are more kinetically favored than the regioisomers 1.4. Our results are in complete agreement with experimental.
Based on the information provided in Table 2, we observe that in this reaction, the cycloadduct CA1 (-25.38 kcal/mol) is thermodynamically more stabilized compared to the contributions of cycloadducts CA2 (-21 kcal/mol), CA3 (-24.73 kcal/mol), and CA4 (-24.25 kcal/mol), respectively. Consequently, we obtain only one cycloadduct observed experimentally.
Table 2
B3LYP/6-31G(d) relative energies (in kcal mol− 1) for the reactants ,TSs and CAs involved in the 13DC between nitrone 1a and alkene 2a in vacuu, and solvent (toluene).
| |
E(u.a) in vacuum
|
E(u.a) in solv
|
∆E (kcal/mol)
|
∆E ( kcal/mol) In solv
|
|
a1
b1
TS1
TS2
TS3
TS4
CA1
CA2
CA3
CA4
|
-768.527882
-193.108433
-961.609358
-961.609594
-961.606878
-961.606886
-961.678890
-961.671264
-961.677899
-961.677942
|
-768.533365
-193.111633
-961.617172
-961.616293
-961.614571
-961.612887
-961.685445
-961.678480
-961.684403
-961.683644
|
--
--
16.915
16.767
18.471
18.466
-21.27
-16.48
-20.64
-20.67
|
--
--
17.46
18.01
19.09
20.15
-25.38
-21.00
-24.73
-24.25
|
When examining the reaction between nitrone a1 and alkene a2, the lengths of the newly formed bonds, C1-C3 and C2-O, in the 1.3 approach (TS1) are 2.11 and 2.17 Å, respectively. Conversely, in the 2.3 approach (TS2), the lengths of the newly formed bonds, C1-O and C2-C3, are 2.11 and 2.09 Å, respectively (see Fig. 2). The lengths of the new bonds formed, C1-C3 and C2-O are 2.082 and 2.168 Å in the 1.3 approach (TS3), while the lengths of the new bonds formed, C1-O and C2-C3, are are 2.082 and 2.074 Å in the 2.3 approach (TS4). Analysis of the geometries at the TS structures given in Fig. 2 shows that the asynchronous bond formation processe for TS1(TS3) is greater compared to that of TS2(TS4).
The concept of bond order (BO) [10] provides a valuable tool for conducting a thorough analysis of the degree of bond formation or breakage throughout a reaction pathway. This theoretical approach has proven effective in studying the molecular mechanism of chemical reactions. The evaluated bond order of C1-C3 and O-C2 bonds that form
bonds at transition states are: 0.414 and 0.321 for TS1 and 0.4389 and 0.3337 for TS3. The evaluated bond order of C2-C3 and O-C1 bonds that form bonds at transition states are: 0.1796 and 0.3679 for TS2 and 0.4193 and 0.3855 for TS4, respectively. Therefore, the asynchronous process of C1-C3 bond formation is more advanced than the O-C2 bond for the TS1 and TS3. While, the asynchronous process of O-C1 bond formation is more advanced than the C2-C3 bond for the TS2. In opposite, the asynchronous process of C2-C3 bond formation is more advanced than the O-C1 bond for the TS4. These results show that this reaction follows an asynchronous concerted mechanism.
The analysis of the electronic properties of this 13DC reaction revealed that the transition state involved the sharing of atomic charges between the dipole 1a and the alkene a2 (see Fig. 2). In the solvent toluene, the charge transfer (CT) in the transitions states, which fluxes from the dipolarophile a2 to the dipole 1a are 0.075, 0.075, 0.075 and 0.075 for TS1, TS2, TS3 and TS4, respectively, Indicating the non-polar nature of this 13DC reaction.
b- Analysis based on local properties.
Domingo proposed a model in 2009 [11] where the formation of a chemical bond is determined by the favorable interaction between an electrophile and a nucleophile. Specifically, the electrophile's most electrophilic site (identified by the highest ωk value) interacts with the nucleophile's most nucleophilic site (identified by the highest Nk value). Figure 3 provides the values of local electrophilicity indices ωk for the O and C3 sites of the dipole, and local nucleophilicity indices Nk for the C1 and C2 sites of the dipolarophile. According to population analysis, the preferred interaction occurs between the C1 site of the alkene and the C3 site of the dipole to give the product a3(1). The analysis based on local properties indicates that regioisomer 1 is more favorable than regioisomer 2. This result is in agreement with the analyses shown by activation barrier calculations.
3.3. Tobologicals analyses
a-Electrostatic Potential (ESP) Analysis
ESP is a potent topological colored map with a variety of active electronic sites in the structural system. The results of this analysis were summarized in Fig. 4 for TS1 of the studied reaction. As the electrostatic potential assigned high electronic density in the blue surface regions, this refers to less tightly located electrons. Additionally, the electronic density was postulated as high to form as rich electronic regions, so these sites can react with electrophiles (nucleophilic attack of O1 on C2). This fact may be attributed to a significant electron withdrawal effect of methylol (CH2OH) substituent, leaving a partial positive charge on C2 reactive site. Based on the ESP map, the most nucleophilic active sites represented in oxygen attached to nitrogen in the five membered ring reactant (a1). The red regions represent the poor-electronic sites appeared in other regions over the system.
b-Reduced Density Gradient / Non-Covalent Interaction (RDG-NCI) analysis
Some important non covalent interactions were generated by the aid of RDG technique accompanied with specified color codes [31]. Figure 5 shows RDG dramatic color change over the system according to sign(λ2) ρ index, where the green spikes describe VdW electrostatic interaction and red spike refers to strong repulsion interactions. H-bond formation is a current state support the stability of generated transition state. Some H-bond character appeared at in the interaction centers. The presence of a significant repulsion interaction in the reactive region indicating delocalization of electronic density emerged from the two reactive parts. This fact mainly is due to other substituents like non-interactive O of (a1) and CH2OH of b1 visualized in Fig. 5a. The spikes appeared in Fig. 5b based on the type of interaction present in the system is mentioned above.
c- Electron localization function
Other supported analysis describing the main path of reaction mechanism, is ELF. This type can predict the electron localized areas on the system surface and then specify the nature of the bond between atoms. Figure 6 Displays two planes (N-C3-C1 and N-O1-C2) of 2D surface map for the formed TS1. As the plane is generated with specifying three successive atoms, the map shows a significant variation in electron localization density with a characteristic color code [32] around the studied 3 atoms plane. In Fig. 3a, the electronic density around the interactive centers, C3 and C1, nearly negligible as the electronic red color parts distributed away from the main centers. While in Fig. 3b, the electronic density less deformed and significantly localized O1 and directed to C2. This discussion confirmed the initial nucleophilic attack step of the partially negative O1 on the partially positive C2.
{kind=link}
{kind=link}
{kind=link}