Materials and methods
All chemicals and also solvents were purchased from Merck, Sigma-Aldrich and Fluka Companies and utilized without further purifications. Melting points were measured by use of an Electrothermal Engineering IA9100 apparatus. 1H and 13C NMR spectra were recorded on a Bruker Avance III 400 MHz spectrometer. The X-ray powder diffraction (XRD) pattern of the catalyst was evaluated using a Philips PW 1830 X-ray diffractometer with CuKα source (λ = 1.5418 Å) in a range of Bragg’s angle (10°–80°) at room temperature. Scanning electron microscope (SEM) analysis was performed by employing VEGA//TESCAN KYKY-EM3200 microscope (acceleration voltage 26 kV). A DuPont 951 Analyzer was exploited for thermogravimetric analysis (TGA) with the heat rate of 10°C/min under N2 atmosphere. CHN analysis was accomplished by means of CHN Elemental Analyzer (LECO 600). Energy Dispersive X ray (EDX) analysis was performed by exerting a Rontec analyzer. Magnetization analysis was carried out using vibration sample magnetometer (VSM, MDK, and Model 7400) at room temperature. IR spectra were recorded on a FT-IR Bruker vector 22 spectrophotometer. Removing the solvents was performed by a Büchi RE-200 Rotary Evaporator.
Preparation of Fe3O4 nanoparticles
Synthesis of Fe3O4 nanoparticles was carried out by co-precipitation method, as reported in the literature [49]. Accordingly, a solution of FeCl3.6H2O (23.3 g, 8.61 mmol) and FeCl2.4H2O (8.60 g, 4.32 mmol) in deionized water (700 mL) was prepared under nitrogen atmosphere at 90 ◦C. Next, 100 mL of ammonium hydroxide (NH4OH 25%) was added dropwise to this solution together with vigorous stirring. As turning the color of the solution to black which represents the generation of Fe3O4, the prepared magnetite nanoparticles were separated by means of an external magnet, washed several times with deionized water and finally dried overnight in a vacuum oven at 100 ◦C. The obtained Fe3O4 nanoparticles were characterized by TGA, XRD, SEM, IR analyses.
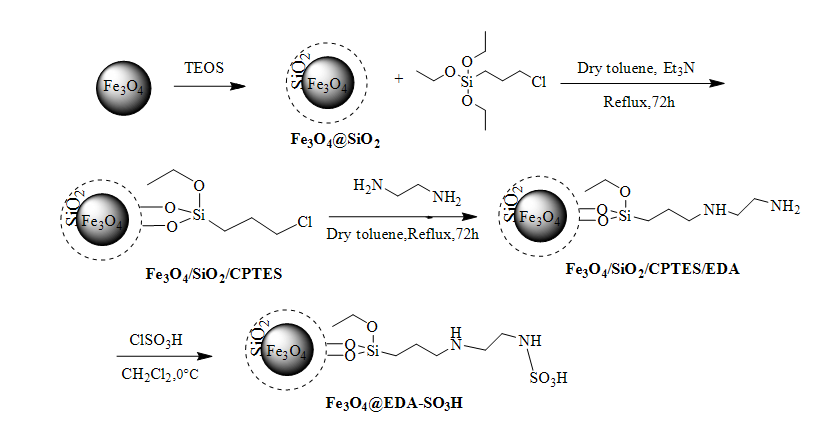
Preparation of Fe3O4/SiO2/CPTES
At first, modification of the Fe3O4 with a silica coating (Fe3O4@SiO2 core-shell) was accomplished according to the literature [50]. In a typical procedure, 1.0 g of the Fe3O4 was dispersed in ethanol (40 mL) and deionized water (10 ml) along with subsequent sonication for 15 min. After that, tetraethyl orthosilicate (TEOS, 1.5 mL) was added to the mixture and subjected to further sonication for 10 min. After sonication, aqueous ammonia (28 %, 1.4 mL) was added in a dropwise manner and the reaction was allowed to stir at 80 °C for 12 h. Then, attained silica coated magnetite nanoparticles were separated from the solution by using an external magnet and after washing several times with distilled water and methanol, were dried at 80 °C under vacuum.
In order to prepare Fe3O4/SiO2/CPTES, a suspension of Fe3O4@SiO2 (1.0 g) in dry toluene (10 mL) was exposed to sonication (for 10 min.). Afterwards, (3-chloropropyl) triethoxysilane (1.5 mL) and triethylamine (0.5 mL) were added and the reaction mixture was stirred under reflux condition for 72 h. Finally, the obtained solid was readily separated by external magnet and dried overnight under vacuum at 60 °C after being washed with ethanol, methanol and toluene.
Preparation of Fe3O4/SiO2/CPTES/EDA
In a two-neck round bottom flask (100 mL) equipped with mechanical stirrer, ethylenediamine (1.0 mL) was added to a suspension of Fe3O4/SiO2/CPTES (1.0 g) in dry toluene (10 mL) and the mixture was stirred for 72 h under reflux condition. Eventually, the brown product (Fe3O4/SiO2/CPTES/EDA) was afforded via separating by external magnet, washing several times with ethanol (10 mL) and drying for 12 h in a vacuum oven at 60 °C.
Synthesis of Fe3O4@EDA-SO3H
A mixture of Fe3O4@EDA (1.0 g) in CH2Cl2 (2 mL) in a two-neck round bottom flask (100 mL) was subjected to sonication for 10 min. Then, the reaction vessel was transferred into an ice bath and cooled to 0 °C. After that, chlorosulfonic acid (1 mL) was added dropwise over 2 h. After completion of the addition, the mixture was stirred at room temperature for 2 h. The resulting solid (Fe3O4@EDA-SO3H) was collected by an external magnet, washed with ethanol (5 mL) and dried at 80 °C for 12 h. The synthesized catalyst was well identified by means of TGA, VSM, CHN, XRD, SEM, IR and EDX analyses.
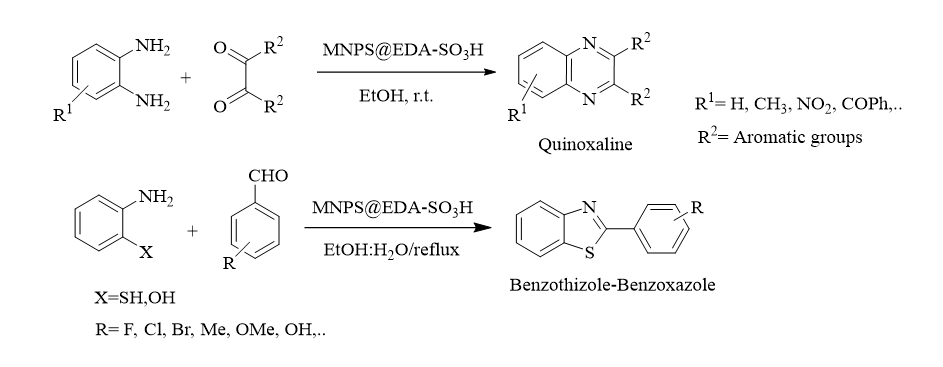
General procedure for the synthesis of quinoxaline derivatives
In a 25 mL two-neck round bottom flask equipped with mechanical stirrer, a mixture of 1,2-diketone (1 mmol), 1,2-phenylenediamine (1 mmol), and Fe3O4@EDA-SO3H (0.032 g) in ethanol (3 mL) were allowed to stir at room temperature. The progress of the reaction was screened by thin-layer chromatography (TLC, n-hexane: ethylacetate 3:1). After completion of the reaction, the catalyst was separated using an external magnet. Then, the solvent was evaporated under reduced pressure and if needed, the desired product was isolated by silica gel column chromatography (n-hexane: ethylacetate). The products were characterized with melting points, 1H and 13CNMR spectroscopy.
General procedure for the synthesis of benzothiazoles and benzoxazoles
To a mixture of 2-aminothiophenol (1 mmol) and aryl aldehyde (1 mmol) in 3 mL of ethanol/water (1:1), Fe3O4@EDA-SO3H (0.032 g) was added and the reaction mixture was stirred at reflux temperature (80 °C) for appropriate time. After completion of the reaction as monitored by TLC, the catalyst was isolated from reaction mixture using a magnet. Next, the solvent was removed under reduced pressure and the crude product was purified by recrystallization from water and washing the obtained solid with ethanol. If needed, silica gel column chromatography (n-hexane: ethylacetate) was exploited for further purification. The products were characterized by 1H, 13CNMR, melting point and comparison with the data reported in the literature.
Spectral data for selected products
3,2-Diphenylquinoxaline(Table 3, entry 1): Yield: 97 %; m.p.: 126-128°C (Lit. [52] 125-126 °C);
1H-NMR(CDCl3, 400 MHz) δ= 8.21(dd, 3J=6.4 Hz, 4J=3.6 Hz, 2H), 7.8(dd, 3J=6.4 Hz, 4J=3.6 Hz, 2H), 7.53-7.56(m, 4H, Ar), 7.28-7.39(m, 6H, Ar). 13C-NMR (CDCl3, 100 MHz): δ= 153.49, 141.24, 139.08, 129.98, 129.85, 129.22, 128.82, 128.29.
Acenaphtho[1,2-b] quinoxaline (Table 3, entry 2): Yield: 96 %; m.p.: 237-240°C (Lit. [51] 237-240 °C); 1H-NMR (CDCl3, 400 MHz): δ (ppm) = 8.42 (d, J=6.8 Hz, 2H), 8.22 (dd, J= 5.2, 3.6 Hz, 2H), 8.10 (d, J=8.4 Hz, 2H), 7.84 (t, J= 7.6 Hz, 2H), 7.77 (dd, J= 6.2, 3.6 Hz, 2H). 13C-NMR (CDCl3, 100 MHz): δ= 154.09, 141.27, 136.50, 131.81, 130.01, 129.60, 129.49, 129.24, 128.68, 121.87.
6-methyl-2,3-diphenyl-quinoxaline (Table 3, entry 3): Yield: 95 %; m.p.: 114-117°C (Lit. [52] 116-117 °C); 1H-NMR (CDCl3, 400 MHz): δ (ppm) = 8.10 (d, J= 8.0 Hz, 1H), 7.98 (s, 1H), 7.63 (dd, J= 8.4, 1.6 Hz, 1H), 7.51-7.54 (m, 4H), 7.28-7.38 (m, 6H), 2.60 (s, 3H). 13C-NMR (CDCl3, 100 MHz): δ= 153.34, 152.59, 141.30, 140.51, 139.72, 139.23, 132.32, 129.85, 129.83, 128.71, 128.70, 128.64, 128.24, 128.03, 21.94.
6- Methyl-snephto[2,1-b]quinoxaline-11-one(Table 3, entry 4): Yield: 96 %; m.p.: 226-228°C (Lit. [51] 228-229 °C); 1H- NMR(CDCl3, 400 MHZ): δ= 8.33(t, J=6.2 HZ, 2H), 8-8.04(m, 3H, Ar), 7.91(s, 1H), 7.77(t, J=7.2 Hz, 2H ), 7.52(d, J=8.4 Hz, 1H), 2.6(s, 3H). 13C-NMR (CDCl3, 100 MHz): δ=153.96, 153.25, 141.16, 139.72, 139.05, 136.18, 131.85, 131.31, 129.93, 129.39, 129.24, 128.98, 128.63, 128.60, 121.73, 121.57, 30.81.
Indeno[2,1-b]quinoxaline-11-one(Table 3, entry 5): Yield: 96 %; m.p.: 217-220°C (Lit. [56] 217-218 °C); 1H-NMR(CDCl3, 400 MHz): δ= 8.24-8.27(m, 1H, Ar), 8.12-8.15(m, 2H, Ar), 7.93-7.96(m, 1H, Ar), 7.82-7.86(m, 1H, Ar), 7.75-7.81(m, 2H, Ar), 7.60-7.64(m, 1H, Ar). 13C-NMR (CDCl3, 100 MHz): δ=189.96, 156.63, 149.28, 143.13, 142.64, 141.55, 136.84, 136.67, 132.53, 132.47, 131.61, 130,30, 129.67, 124.80, 122.53.
Phenantro[9,10-b]quinoxaline-11-one(Table 3, entry 6): Yield: 98 %; m.p.: 220-223°C (Lit. [51] 223-225 °C); 1H-NMR(CDCl3, 400 MHZ): δ=9.41(d, J=7.6 HZ, 2H), 8.57(d, J=8 Hz, 2H), 8.33-8.34(m, 2H, Ar), 7.82-7.87(m, 2H, Ar), 7.74-7.81(m, 2H, Ar). 13C-NMR (CDCl3, 100 MHz): δ= 142.44, 142.18, 132.05, 130.30, 129.75, 129.45, 127.93, 126.26, 122.91.
2-phenyl-benzothiazole (Table 6, entry 1): Yield: 97 %; m.p.: 112–114 °C (Lit. [53] 112-113 °C); 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 8.24-8.25 (m, 1H, Ar), 8.16-8.18 (m, 1H, Ar), 8.05-8.10 (m, 2H, Ar), 7.77-7.79 (m, 1H, Ar), 7.47-7.59 (m, 4H, Ar). 13C-NMR (DMSO-d6, 100 MHz): δ= 165.94, 153.79, 135.38, 135.09, 134.46, 132.07, 129.64, 127.33, 126.89, 126.37, 123.58, 123.02, 122.95.
4-(benzo[d]thiazol-2-yl)benzaldehyde (Table 6, entry 3): Yield: 90 %; m.p.: 133 °C (Lit. [54] 132-133 °C); 1H-NMR (CDCl3, 400 MHz): δ (ppm) = 10.09 (s,1H,CH), 8.26 (d, J= 8.0 Hz, 1H, Ar), 8.17-8.21 (m,1H, Ar), 8.12 (dd, J= 7.6, 3.2 Hz, 1H, Ar), 7.99-8.05 (m, 2H, Ar), 7.94 (dd, J= 8.0, 4.4 Hz, 1H, Ar), 7.51-7.56 (m, 1H, Ar), 7.42-7.46 (m, 1H, Ar). 13C-NMR (CDCl3, 100MHz): δ=191.51, 130.29, 128.07, 128.04, 126.74, 126.57, 125.92, 125.58, 123.72, 123.44, 121.78, 121.71.
2-(4-chlorophenyl)benzoxazole (Table 6, entry 16): Yield: 95 %; m.p.: 133 °C (Lit. [55] 144-148 °C); 1H-NMR (DMSO-d6, 400 MHz): δ (ppm) = 7.87 (d, J= 8.0 Hz, 2H), 7.48 (d, J= 8.4 Hz, 2H), 7.32(d, J= 8.0 Hz, 1H), 7.28-7.21 (m, 1H), 7.04 (d, J= 8.0 Hz, 1H), 6.95-6.91 (m, 1H). 13C-NMR (DMSO-d6, 100MHz): δ=165.5, 152.4, 137.7, 135.1, 134.3, 129.9, 129.3, 129.2, 120.17, 115.8, 115.2.
{kind=link}
{kind=link}