Resource availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yingying Pu ([email protected]).
Materials availability
Plasmids generated in this study are available from the lead contact upon request.
Data and code availability
Sequencing data, the processing data and original code have been deposited to GEO repository (Uploading).
Bacterial strains and growth conditions
The bacterial strains used in this study included Escherichia coli strains MG1655, Caulobacter crescentus NA1000 and Staphylococcus aureus strain ATCC 25923 were included. Cultures of E. coli were grown in Luria Broth (LB) medium. Caulobacter crescentus strain NA1000 was grown in peptone yeast extract (PYE) medium. And Staphylococcus aureus strain ATCC 25923 was grown in Mueller-Hinton Broth (MHB) medium. All bacterial strains were routinely grown at 37°C and 220 rpm. To maintain plasmids, when necessary, media were supplemented with chloramphenicol (25 µg/ml). For arabinose-induction system expression experiments, 0.002% arabinose was supplemented in the medium.
Strains construction. The construction of recombinant plasmids was performed using the 2 × MultiF Seamless Assembly Mix (ABclonal, RK21020). To label the PdeI gene, pdeI, along with its native promoter, was fused with either gfp or bfp and cloned into the pBAD backbone. For gene overexpression, the target genes of interest were cloned into either a p15A ori plasmid or a pUC ori plasmid, under the control of the Arabinose-induction system. For the c-di-GMP sensor (Snapgene: #182291), the plasmid origin was replaced with the p15A ori.
RiboD -PETRI
Cell preparation. E. coli MG1655 cells were cultured overnight and subsequently diluted at a ratio of 1:100 into fresh LB medium and grown statically for 24h at 37°C.For 3h exponential period E. coli sample, E. coli MG1655 cells were grown overnight and then diluted 1:100 into fresh LB medium and grown for 3 h at 37℃ and 220 rpm. Caulobacter crescentus strain NA1000 cells were grown overnight and then diluted 1:100 into fresh Mueller-Hinton Broth (MHB) medium and grown for 9h at 37℃ and 220 rpm. And Staphylococcus aureus strain ATCC 25923 cells were grown overnight and then diluted 1:100 into fresh peptone yeast extract (PYE) medium and grown for 3 h at 37℃ and 220 rpm. All the culture was vigorously shaken using a vortex, and the cells were then centrifuged at 5,000g for 2 minutes at 4°C. The pellet was resuspended in 2 ml of ice-cold 4% formaldehyde (F8775, Millipore Sigma, diluted into PBS). This suspension was rotated at 4°C for 16 hours.
Cell permeabilization. 1 ml of fixed cells were centrifuged at 5,000g for 5 minuts at 4°C, followed by resuspension in 1 ml washing buffer (100 mM Tris-HCl pH7.0, 0.02 U/μl SUPERase-In RNase Inhibitor, AM2696, Invitrogen). After another centrifugation at 5,000g for 5 minuts at 4°C, the supernatant was removed. The pellet was then resuspended in 250 μl permeabilization buffer (0.04% Tween-20 in PBS-RI, PBS with 0.01 U/μl SUPERase-In RNase Inhibitor) and incubated on ice for 3 minuts. 1 ml cold PBS-RI was added, and the cells were centrifuged at 5,000g for 5 minuts at 4°C, followed by resuspension in 250 μl Lysozyme Mix (Dissolving 250 μg/ml Lysozyme or 5 μg/ml Lysostaphin for S. aureus in TEL-RI buffer, comprising 100 mM Tris, pH 8.0 (AM9856, Invitrogen), 50 mM EDTA (AM9261, Invitrogen), and 0.1 U/μl SUPERase In RNase Inhibitor). The samples were incubated at 37°C and mixed gently every minute. Then cold PBS-RI (1ml) was added immediately, and cells were centrifuged at 5,000g for 5 min at 4°C. The cells underwent another wash with 1 ml cold PBS-RI. Subsequently, cells were resuspended in 40 μl DNaseI-RI buffer (4.4 μl 10×reaction buffer, 0.2 μl SUPERase In RNase inhibitor, 35.4 μl H2O), followed by addition of 4 μl DNaseI (AMPD1, Millipore Sigma). The samples were incubated for 30 min at room temperature and mixed gently every 5 minutes. 4 μl Stop Solution was added, and the samples were incubated for 10 minutes at 50°C with gentle mixing every minuts. Following centrifugation at 5,000g for 10 minutes at 4°C, cells were washed twice with 0.5 ml cold PBS-RI. Finally, cells were resuspended in 200 μl cold PBS-RI, and their count and integrity were assessed using the ACEA NovoCyte flow cytometer with a 100× oil immersion lens.
Primer preparation. For round 1 RT, round 2 and round 3 ligation reactions, all primers design and preparation as previously described 7. All primers were purchased from Sangon Biotech (Table S1). For ligation primers preparation, mixtures were prepared as follows: 31.1 μl each R2 primer (100 μM), 28.5 μl SB83 (100 μM) and 21.4 μl H2O, were splitted to 2.24 μl for one sample. Mixtures containing 63.2 μl each R3 primer (70 μM) and 58 μl SB8 (70 μM), were splitted to 3.49 μl for one sample. Before use, ligation primers were incubated as follows: 95 °C for 3 min, then decreasing the temperature to 20 °C at a ramp speed of −0.1 °C s−1, 37 °C for 30 min. For blocking mix preparation, 50 μl primer SB84 (400 μM) and 80 μl primer SB81 (400 μM) were incubated as follows: 94°C for 3 min, then decreasing the temperature to 25 °C at a ramp speed of −0.1 °C s−1, 4°C for keeping. Round 2 blocking primers were mixed as follows: 37.5 μl 400 μM SB84, 37.5 μl 400 μM SB85, 25 μl 10× T4 ligase buffer, 150 μl H2O. Round 3 blocking primers were mixed as follows: 72 μl 400 μM SB81, 72 μl 400 μM SB82, 120 μl 10× T4 ligase buffer, 336 μl H2O, 600 μL 0.5 M EDTA.
Round 1 RT reaction. About 3×107 cells were introduced into an RT reaction mix composed of 240 μl 5× RT buffer, 24 μl dNTPs (N0447L, NEB), 12 μl SUPERase In RNase Inhibitor and 24 μl Maxima H Minus Reverse Transcriptase (EP0753, Thermo Fisher Scientific). Nuclease-free water was added to achieve a total reaction volume of 960 μl, and the mixture was thoroughly mixed by vortexing. Subsequently, 8 μl of the reaction mixture was dispensed into each well of a 96-well plate, where 2 μl of each RT primer had been added previously. The sealed 96-well plate was inverted repeatedly for thorough mixing, followed by a brief spin. The plate was then incubated as follows: 50 °C for 10 min, 8 °C for 12 s, 15 °C for 45 s, 20 °C for 45 s, 30 °C for 30 s, 42 °C for 6 min, 50 °C for 16 min, and finally held at 4 °C. After the RT process, all 96 reactions were pooled into one tube. 75 μl of 0.5% Tween-20 was added, and the reactions were incubated on ice for 3 minutes. Cells were centrifuged at 7,000g for 10 minutes at 4°C and then resuspended in 0.4 ml PBS-RI. Thirty-two microliters of 0.5% Tween-20 was added, and the cells underwent centrifugation at 7,000g for 10 minutes at 4°C.
Round 2 ligation reaction. Cells were resuspended in 500 μl 1× T4 ligase buffer, followed by the addition of 107.5 μl PEG8000, 37.5 μl 10× T4 ligase buffer, 16.7 μl SUPERase In RNase Inhibitor, 5.6 μl BSA, and 27.9 μl T4 ligase (M0202L, NEB). The reaction solution was thoroughly mixed by vortexing. Subsequently, 5.76 μl of the reaction mixture was dispensed into each well of a 96-well plate, where 2.24 μl of each round 2 ligation primer had been added previously. The sealed 96-well plate was inverted repeatedly for thorough mixing and then subjected to a short spin. The plate was incubated at 37°C for 45 minutes. Following this, 2 μl of round 2 blocking mix was added to each well and incubated at 37°C for an additional 45 minutes. All 96 reactions were pooled into one tube after incubation.
Round 3 ligation reaction. A mixture comprising 89 μl H2O, 26 μl PEG8000, 46 μl 10× T4 ligase buffer, and 12.65 μl T4 ligase was prepared and thoroughly mixed by vortexing. Subsequently, 8.51 μl of the reaction mixture was dispensed into each well of a 96-well plate, where 3.49 μl of each round 3 ligation primer had been added previously. The sealed 96-well plate was inverted repeatedly for thorough mixing and then subjected to a brief spin. The plate was incubated at 37°C for 45 minutes. Following this, 10 μl of round 3 blocking mix was added to each well and incubated at 37°C for an additional 45 minutes. All 96 reactions were combined into one tube after incubation.
Cells lysis. 42 μl of 0.5% Tween-20 was added, and cells were centrifuged at 7,000g for 10 minutes at 4°C. The cells underwent two washes using 200 μl TEL-RI containing 0.01% Tween-20, each time centrifuged at 7,000g for 10 minutes at 4°C. Subsequently, cells were resuspended in 30 μl TEL-RI buffer. Cell counting and integrity checks were performed using the ACEA NovoCyte flow cytometer with a 100× oil immersion lens. A moderate amount of cells was then added to the lysis buffer (50 mM Tris pH 8.0, 25 mM EDTA, 200 mM NaCl, 0.5% Triton X-100), and 5 μl of proteinase K (AM2548, Invitrogen) was introduced. Samples were incubated at 55°C for 60 minutes and gently mixed every minute.
Library construction. To facilitate template switching, lysates were purified with VAHTS DNA Clean Beads (N411, Vazyme) at a ratio of 2.0×, and cDNA was eluted in 12 μl of water. The purified cDNA was then combined with 4 μl of 5× RT buffer, 1 μl of dNTPs (N0447L, NEB), 0.5 μl of SUPERase In RNase Inhibitor, 0.5 μl of Maxima H Minus Reverse Transcriptase, and 0.5 μl of the TSO primer (100 mM, Table S1). This reaction solution underwent incubation as follows: 25°C for 30 min, 42°C for 90 min, 85°C for 5 min, and then held at 4°C. Subsequently, 1 μl of RNaseH was added, and the reaction solution was incubated at 37°C for 30 min. The cDNA was purified once again with VAHTS DNA Clean Beads at a ratio of 2.0× and eluted in 13 μl of H2O. The integrity of the cDNA was assessed using primers TSO-2 and R1 or R2 or R3 by qPCR (Table S1).
Ribosomal RNA derived cDNA depletion (RiboD). We developed a set of cDNA probe primers to selectively deplete r-cDNA (Table S1). These probe primers possess the ability to specifically hybridize with r-cDNA and also hybridize with a biotin-labeled universal primer. In the reaction, 5 μl of r-cDNA probe primers (10 μM), 2.5 μl of 10× hybridization buffer (Tris-HCl pH 8.0 100 mM, NaCl 500 mM, EDTA pH 8.0 10 mM), and 5 μl of biotin primer (10 μM) were added to 12.5 μl of purified cDNA. The reaction solution underwent incubation as follows: 95°C for 2 min, followed by a temperature decrease to 20°C at a ramp speed of −0.1°C s−1, and then held at 37°C for 30 min. Subsequently, 20 μl of Streptavidin magnetic beads (BEAVER, 22307) was washed twice using 1 ml of 1× B&W buffer (Tris-HCl pH 7.5 10 mM, EDTA 1 mM, NaCl 1M, Tween-20 0.05%) and resuspended in 25 μl of 2× B&W buffer. Twenty-five microliters of washed Streptavidin magnetic beads were added to 25 μl of annealed cDNA. The reaction solution was incubated at room temperature for 30 min with gentle mixing per minute. Following this, the reaction solution tube was placed into a magnetic stand to collect the supernatant. The cDNA depleted of r-cDNA was purified using VAHTS DNA Clean Beads at a ratio of 2.0× and eluted in 12.5 μl of H2O. The depletion of r-cDNA could be repeated using the above protocol, and ultimately, the cDNA was eluted in 20 μl of H2O.
Library amplification and sequencing. To the 20 μl cDNA solution, the following components were added: 2.4 μl R3 primer (10 mM, Table S1), 2.4 μl TSO-2 primer (10 mM, Table S1), 40 μl 2× KAPA HIFI mix (KAPA, 2602), 1.6 μl Sybr green (25×), 0.8 μl MgCl2 (0.1 M), and 12.8 μl H2O. This PCR reaction solution was placed in a thermocycler and incubated with the following parameters: 98 °C for 45 s, followed by cycling of 98 °C for 15 s, 60 °C for 30 s, and 72 °C for 60 s. Cycling continued on a qPCR machine until the reaction approached saturation. PCR products were then purified using VAHTS DNA Clean Beads at a ratio of 0.9× and eluted in 25 μl of H2O. Finally, the purified PCR products underwent end repair and adaptor ligation using the VAHTS Universal DNA Library Prep Kit for Illumina V3 (Vazyme, ND607).
Bulk RNA-seq library construction
Total RNA of the samples was extracted utilizing the Bacteria RNA Extraction Kit (R403-01, Vazyme). Subsequently, the RNA underwent mRNA enrichment (N407, Vazyme), fragmentation, cDNA synthesis, and library preparation using the VAHTSTM Total RNA-seq (H/M/R) Library Prep Kit for Illumina® (NR603, Vazyme).
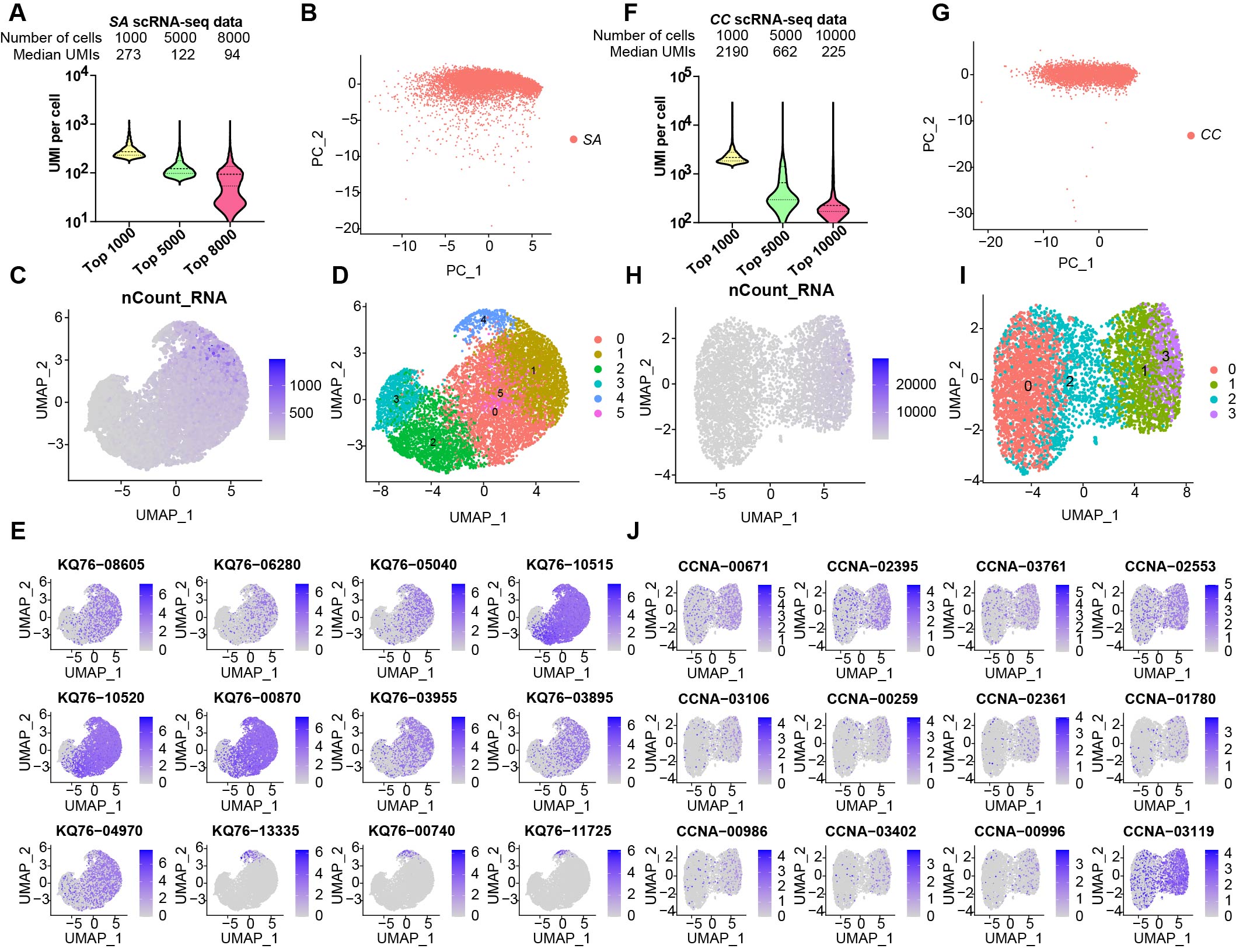
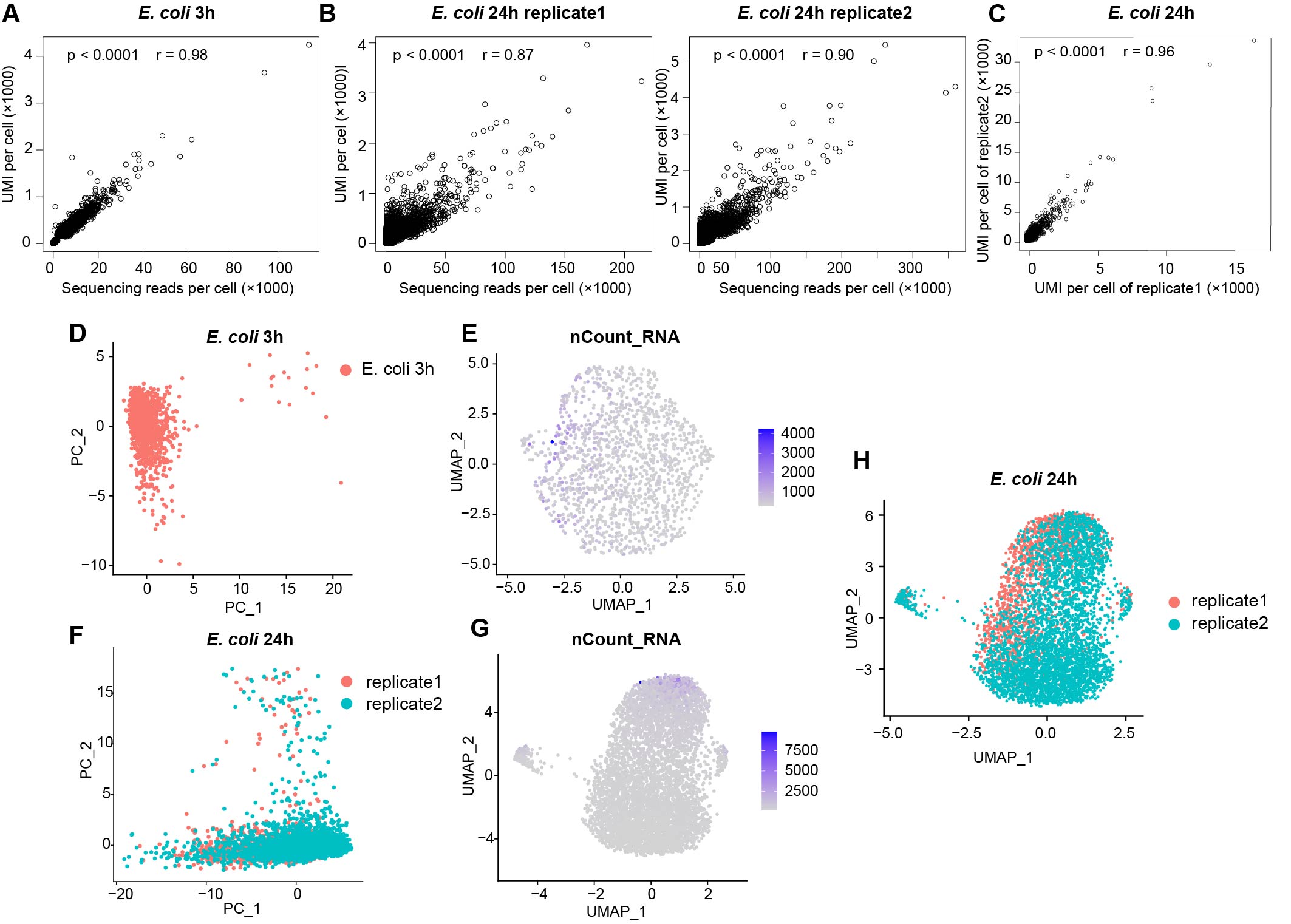
Bioinformatics analysis Methods
Single-Cell Analysis. The sequencing data underwent processing into matrices using scripts and a pipeline as previously described7 in Python 2.7.15, with some modifications (the detailed original code and all the data were deposited in the GEO repository). After the count tables were made, subsequent analysis of single-cell data was conducted using Seurat package (version 4.3.0; http://satijalab.org/seurat/) in R (https://www.r-project.org/). Since there are two replicates of E coli static biofilm, these two datas need to be merged into one SeuratObject and removed batch effects. However, the samples for exponential period E. coli, S. aureus and C. crescentus only had one sample, so they did not need this process. At the beginning of doing the scRNA-seq analysis, we screened the datas of all samples. For preprocessing of E coli static biofilm data, cells were filtered with UMI per cell more than 100 and less than 2000 for replicate 1 and replicate 2 to obtain 1621 and 3999 cells, respectively. For exponential period E coli data. The data was screened for cells with UMIs greater than 200 and less than 5000 to obtain 1464 cells. The screening criteria of S. aureus were cells with UMIs greater than 15 and less than 1000 and genes greater than 30 (1000>UMIs>15, gene counts>30). The screening criteria of C. crescentus were cells with UMIs greater than 200 and less than 5000 and gene counts greater than 30 (5000>UMIs>200, gene counts>30). After screening, all the datas were normalized using a scale factor of 10000 through a global-scaling normalization method called "LogNormalize". Highly variable features were then identified, returning 500 features per dataset. Then we combine the data of the two replicates of E coli static biofilm into a single SeuratObject by FindIntegrationAnchors and IntegrateData functions. Then all the datas underwent scaling using the ScaleData function, followed by dimension reduction through principal component analysis. After principal component analysis, we removed batch effect by RunHarmony of the two replicates of E coli static biofilm datas. Then a graph-based clustering approach was employed in all datas to identify clusters of gene expression programs using the Louvain algorithm (Seurat 4.3.0). The dims we choosed were 6. And the resolution was 0.3 for C. crescentus and S. aureus or 0.4 for E coli datas. Marker genes for each cluster were computed using the Wilcoxon Rank-sum test. Specifically, marker genes for each cluster were initially obtained using the FindMarkers function of Seurat. Then we performed pathway enrichment analysis of marker gene by clusterProfiler function16 within R.
Comparison of scRNA-seq with Bulk RNA-Seq. The bulk RNA-seq clean data reads were mapped to the E coli MG1655 k12 genome (EnsemblBacteria Taxonomy ID: 511145) using the BWA aligner software (v0.7.17-r1188, https://github.com/lh3/bwa.git). Converting sam files to bam files using samtool (v1.9). The mapping results were counted by featureCounts (v2.0.1, https://github.com/topics/featurecounts) to generate expression results. Single-cell and bulk transcriptomes of E coli were compared by computing the Pearson correlation of log2 reads per gene of bulk RNA-seq and log2 UMI per gene of scRNA-seq.
Flow cytometry and FACS analysis
All samples were measured on a Beckman CytoFLEX SRT flow cytometer with a 70 μm nozzle in which normal saline was used as sheath fluid. PdeI-BFP or c-di-GMP sensor labeled strain in the 24 h static biofilm growth phase was washed and resuspended in sterile PBS. Microorganisms were identified by FSC (forward scatter) and SSC (side scatter) parameters. Cells were sorted into different groups based on their fluorescence intensity (PB450 for BFP, FITC for mVenusNB, ECD for mScarlet-I). The results were analyzed by FlowJo V10 software (Treestar, Inc.).
Antibiotic killing and persister counting assay
Cells sorted from FACS were suspended in fresh LB broth containing 150 μg/ml ampicillin and incubated for an additional 3 hours at 37°C with continuous shaking at 220 rpm. Following this, the cells were plated on LB plates for colony-forming unit (CFU) counts before the ampicillin challenge, and the plates were incubated overnight at 37°C. After the ampicillin challenge, cells were washed with PBS buffer and plated again for CFU counts. The persister ratio was defined as the ratio of CFU after ampicillin challenge to CFU before ampicillin challenge. Averages and standard deviations presented are representative of three biological replicates.
Microscopy
Bright-field and fluorescence imaging. Inverted microscopes (Nikon Eclipse Ti2 and Leica Stellaris 5 WLL) were utilized, with illumination provided by different lasers: 405 nm for BFP, 488 nm for GFP, respectively. The fluorescence emission signal was captured by an sCMOS camera (pco.edge 4.2 bi). Filter sets tailored to each fluorophore's spectrum were employed. Image analysis was conducted using ImageJ software (Fiji). For c-di-GMP sensor analysis, the ratio of mVenusNB to mScarlet-I (R) exhibited a negative correlation with the concentration of c-di-GMP. Therefore, R-1 displayed a positive correlation with the concentration of c-di-GMP.
Time-lapse imaging. To observe antibiotic killing and bacteria resuscitation processes, cells labeled with PdeI-GFP in the 24-hour static growth phase were collected, washed twice with PBS buffer, and imaged on a gel pad containing 3% low melting temperature agarose in PBS [REF]. The gel pad was prepared in the center of the FCS3 chamber as a gel island. The cells were then observed under bright field or epifluorescence illumination. Subsequently, the gel pad was surrounded by LB broth containing 150 μg/ml ampicillin for 6 hours at 35°C. Fresh LB broth was flushed in, and the growth medium was refreshed every 3 hours, allowing cells to recover sufficiently.
Determination of c-di-GMP concentration by HPLC-MS/MS
Determination of c-di-GMP concentration by HPLC-MS/MS involved growing MG1655 Δara pBAD::pdeI and MG1655 Δara to mid-exponential growth phase, followed by induction with 0.002% arabinose. After 2 hours of incubation, cells were harvested and washed with PBS. The washed cells were rapidly frozen using liquid nitrogen. Simultaneously, another portion of washed cells was stained with SYTOTM24 and quantified using flow cytometry. c-di-GMP concentration detection was conducted by Wuhan Lixinheng Technology Co. Ltd. through high-pressure liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). All strains were assayed in biological triplicates, and measured values were converted into intracellular c-di-GMP concentrations (pg) per cell.
Quantification and statistical analysis
Statistical analysis was conducted using GraphPad Prism 9 software for Windows. Significance was determined by a two-tailed Student’s t-test. Error bars indicate the standard deviations of the mean from a minimum of three independent experiments. A significance threshold of P < 0.05 was applied. Asterisks were used to denote significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
{kind=link}
{kind=link}
{kind=link}
{kind=link}