Tissue collection and primary cell extraction
Placental chorionic tissues were collected from patients with EOPE after cesarean section (16 cases) and from healthy pregnant women who underwent cesarean section due to nonorganic diseases such as large fetus, small pelvis and scarred uterus (16 cases), and the collection site was approximately 3 cm from the placental cord, part of which was fixed with paraformaldehyde for IHC. Inclusion criteria were defined according to the American College of Obstetricians and Gynecologists (ACOG): PE was defined as hypertension with proteinuria occurring after 20 weeks of gestation. EOPE was defined as PE that developed prior to 34 weeks of gestation. The age of these patients was 26–37 years. Patients with smoking or multiple pregnancies, fetal structural or genetic abnormalities, maternal infections and any other confounding pathology (diabetes mellitus, renal disease, chronic hypertension, hyperthyroidism and hypothyroidism) were excluded. The remaining samples were snap-frozen in liquid nitrogen using lyophilization tubes and stored in a liquid nitrogen tank for RNA and protein extraction. A 10-cm umbilical cord from the placental remnant of a healthy woman delivered by full-term cesarean section was selected for extraction of primary HUVECs for subsequent experiments using a previously reported method[6]. Informed consent was obtained with approval from the Medical Ethics Committee of the Xiangya Hospital Central South University (Ethics number: 202307152). All patients signed an informed consent form before enrollment in the study.
Cell culture
This study used primary HUVECs, which were isolated from freshly obtained human umbilical cords of healthy mothers, as a model of vascular ECs in vitro. The umbilical cords were washed inside and outside with saline in a sterile station, and type I collagenase (0.1%; Solarbio, China) was digested at 37°C for 10 min before addition of a terminating digestion solution (ECM complete medium: 10% fetal bovine serum, 1% epidermal growth factor) and 5 ml of saline rinse. The rinse solution was then centrifuged at 1200 rpm for 5 min to separate the HUVECs from the collagenase, and the HUVECs were resuspended in ECM after measuring the concentration. Cells were seeded at the appropriate concentration in culture flasks and incubated at 37°C in a 5% CO2 incubator. Primary ECs were cultured to the third generation (stable cell state) and then used for experiments.
Real-time quantitative PCR
Total RNA was extracted from cells and tissue using the Steadypure RNA Extraction Kit (Accurate Biotechnology, Hunan). The cDNA templates of miRNAs were synthesized with an Evo M-MLV RT Mix Kit (Accurate Biotechnology, Hunan), while miRNA was synthesized with TransScript miRNA First-Strand cDNA Synthesis SuperMix (+ Dye I/+Dye II). RT‒qPCR was performed using a SYBR Green Premix Pro Taq Hs qPCR Kit (Accurate Biotechnology, Hunan) for miRNA and PerfectStart Green qPCR SuperMix (TransGen Biotech, Beijing) for miRNA. Quantitative real-time PCR was performed using a QuantStudioTM5 system (Applied Biosystems, CA, USA) with a preset PCR program. The primer sequences purchased from Tsingke Biotechnology (Changsha, China) for real-time fluorescent quantitative PCR are shown in Table S1.
Protein extraction and immunoblotting
Protein extraction from tissues and cells was performed using RIPA lysis buffer (Bioss, China), and the concentration was determined using a BCA protein assay kit (Bioss, China). The protein samples were incubated for 5 min at 95°C; then, proteins (20 mg) were subjected to 8% SDS‒PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (0.45 mm pore size; Millipore, MA, USA). The primary antibodies used were rabbit anti-CPT1A (15184-1-AP) and rabbit anti-β-actin from Proteintech. All primary antibodies were used at a 1:1,000 dilution, with secondary HRP antibodies (Abiowell, China) used at 1:8000. Protein strips were developed using an ImageQuant 800 instrument and analyzed by ImageJ.
IHC
Placental tissues were embedded in paraffin. Sections were dewaxed in water, and antigen repair was performed in a microwave oven in a repair cassette filled with citrate antigen repair buffer (pH 6.0) in a microwave oven, washed and blocked for endogenous peroxidase. After serum blocking, a primary antibody against CPT1A (1:300, 15184-1-AP, Proteintech) was added and incubated overnight at 4°C. After incubation with secondary antibodies, the staining was repeated with DAB and hematoxylin.
Subcellular fractionation assay
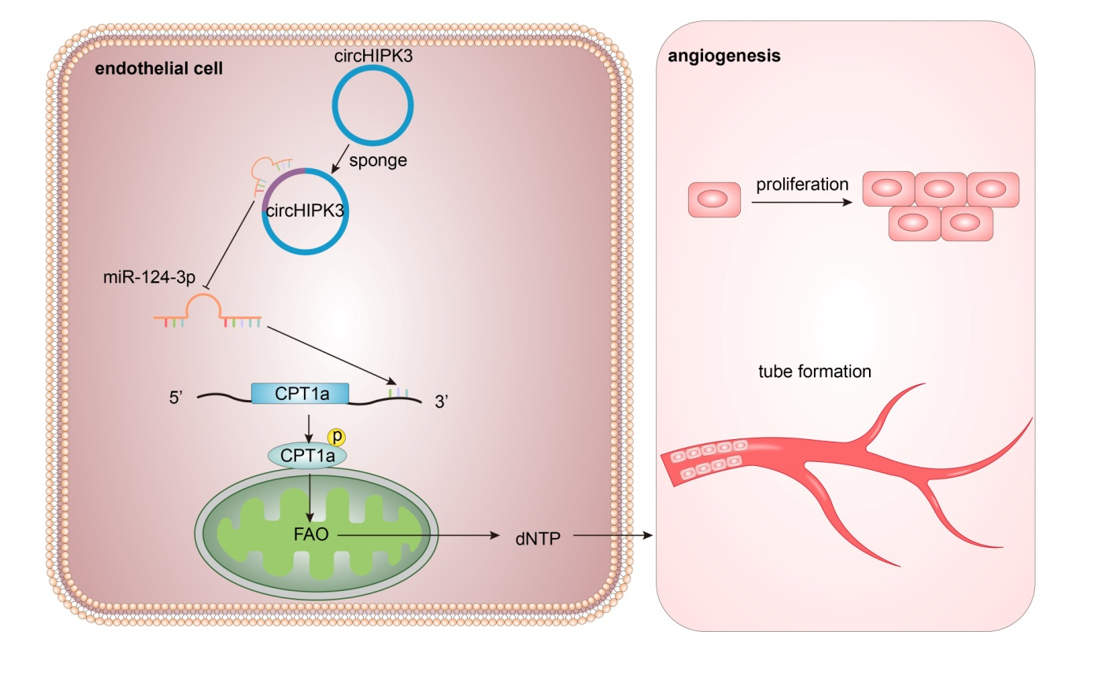
RNA isolated from the cytoplasmic and nuclear extracts obtained from HUVECs by an Invitrogen PARIS nuclear and cytoplasmic extraction kit (AM1921, Invitrogen, Thermo Fisher Scientific) was subjected to RT‒qPCR analysis; see the previous section for specific steps. The levels of U6 (nuclear control), GAPDH (cytoplasmic control), circHIPK and miR-124-3p were determined.
Cell transfection
The pcDNA3.1+/BamHI EcoRI vector (GenePharma, Shanghai, China) was used for CPT1A overexpression, the empty plasmid vector was used as a control, the pGPU7/GFP/Neo vector (GenePharma, Shanghai, China) was used for CPT1A silencing, and sh-NC was used as the NC. GenePharma designed and synthesized four sh-RNAs targeting the CPT1A gene and two sh-RNAs targeting the circHIPK3 gene. sh-CPT1A (sh-941) and sh-circHIPK3 (sh-1), which were identified as the most effective by real-time fluorescence quantitative PCR, were used for further experiments. MiR-124-3p overexpression and repression were achieved using miR-124-3p mimics and miR-124-3p inhibitors, respectively, with NC mimics and NC inhibitors serving as controls (GenePharma). These plasmids were transfected into cells using Lipofectamine 3000 reagent (Thermo Fisher Scientific, Waltham, MA, USA) as needed. Cells were transfected for 48 h and then collected for subsequent analysis.
FAO assay
The FAO assay followed the manufacturer's instructions for the Fatty Acid Oxidation Assay Kit (ab217602, Abcam). A total of 3×104 cells per well were seeded in a 96-well plate and cultured overnight. Then, the cells were rinsed twice with 100 µl of prewarmed FA-free medium and mixed with 90 µl of prewarmed FA measurement medium. The cell-free wells were used as signal controls. Wells were supplemented with 85 µl of FA-free measurement medium and 5 µl of BSA control as the FA-free control. Each well was spiked with 10 µl of extracellular O2 consumption reagent from the Extracellular Oxygen Consumption Assay kit (ab197243, Abcam) except for the blank control. The FAO activator FCCP (0.5 µM) and the inhibitor etomoxir (40 µM) were used as positive and negative controls, respectively, and the concentrations were used as recommended in the instructions and according to a previously described protocol[38]. The wells were sealed with two drops of prewarmed mineral oil, and the fluorescence signal was immediately detected using an instrument (Biotek, Cytation5).
Cell proliferation assay
Cell proliferation assays were performed using CCK-8 assays (K101, APE). Transfected cells were seeded in 96-well plates at a density of 3\(\times\)103 cells/100 ml per well. Proliferation levels were measured at 0, 24, 48 and 72 h post-transfection. CCK-8 solution (10 ml) was added to each well and then incubated for 1 h at 37°C in the dark. The absorbance at 450 nm was measured using a multimode reader (Infinite M200 Pro; Tecan).
Flow cytometry analysis of the cell cycle
The transfected cells were collected by digestion, fixed and stained according to the protocol of the cell cycle assay kit (E-CK-A351, Elabscience) and then detected by flow cytometry (DxpAthena Cytek, USA). The percentages of cells in the G0/G1, S, and G2/M phases were calculated using ModFit software.
Cell migration assay
A Transwell 24-well filter insert (Corning, 8.0 µm pore size) was used to investigate cell migration. In each well, transfected HUVECs (5x104 cells/well) were resuspended in 200 ml of serum-free ECM and placed in the upper chamber, and 500 ml of complete medium was added to the lower chamber. Transwell units were incubated for 24 h at 37°C; the chambers were fixed with 4% paraformaldehyde at room temperature for 30 min and stained with 0.2% crystal violet for 20 min at 37°C. Five areas were randomly selected for counting under an inverted microscope (Leica, DM4B).
Tube-formation assay
The Matrigel gel was melted overnight at 4°C in advance, and the 96-well plates and tips were precooled 2 h in advance. Sixty microliters of Matrigel was added to each well of 96-well plates and incubated in a 37°C incubator for 0.5 h. HUVECs (1.5 X 104 cells/well) were resuspended in 100 ml of conditioned medium and then seeded in wells. After incubation for 3–6 h at 37°C, tube-like structures formed and were imaged using a microscope (100x magnification; Olympus). Photomicrographs from each well were captured, and the total length of tubes was analyzed using ImageJ software (NIH, Bethesda, MD, USA).
Luciferase reporter assay
CPT1A or circHIPK3 containing the miR-124-3p putative binding site was cloned into the pmirGlo vector (GenePharma). Cells were plated in 24-well plates at a density of 5*105 cells per well 1 day prior to transfection. Cells were cotransfected with WT or MUT luciferase vector and miR-124-3p or control using Lipofectamine 3000 reagent (Thermo Fisher Scientific). After 48 h of incubation, a Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) and Multifunctional Enzyme Labeler (Synergy HTX, USA) were used.
Statistics
All data are presented as the mean ± SD. Statistical analyses were performed using GraphPad Prism 9.4.1 software. Unpaired Student’s t test was used to determine the significance of differences between two groups, and one-way ANOVA was used for multiple groups. Statistical significance was defined as P < 0.05. All data were obtained from > 3 independent experiments.
{kind=link}