The landscape of m6A regulators
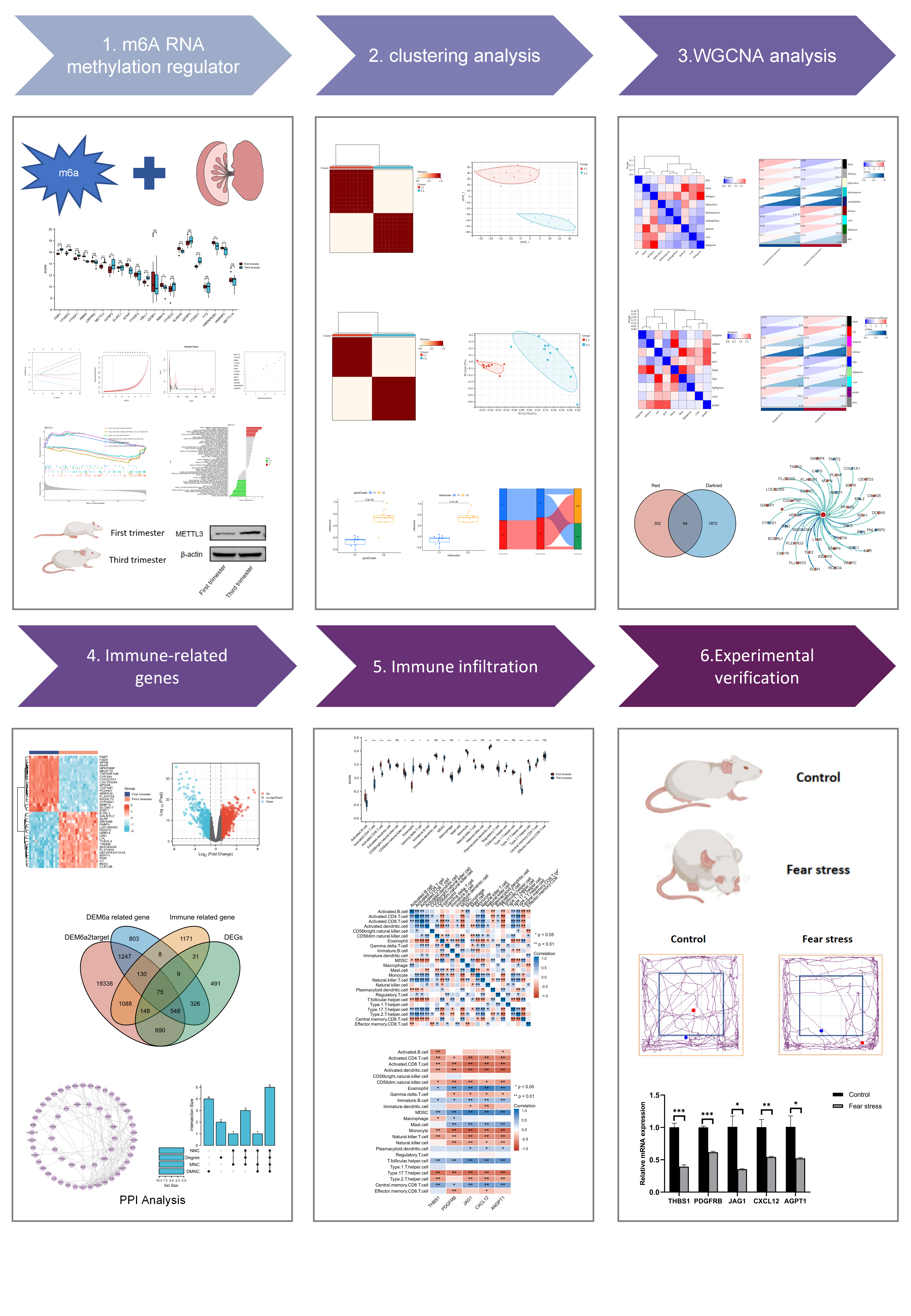
In this study, we curated and analyzed a set of 21 recognized m6A regulators, which comprised 14 writers, 5 readers, and 2 erasers. Through expression profiling, we identified 13 differentially expressed m6A regulators (DEm6AR) that showed significant variation between the first and third trimesters (Fig. 1A). Of these, seven genes (FMR1, YTHDF2, YTHDF2, METTL3, IGFBP2, CBLL1, and YTHDC1) were upregulated, while six genes (RBMX, WTAP, RBM15, ALKBH5, HNRNPA2B1, and HNRNPC) were downregulated. To explore the transcriptome relationships among the m6A regulators, we conducted correlation analysis and found a close association between writers, readers, and erasers (Fig. 1B).
Machine learning
Seven potential diagnostic biomarkers were identified using the LASSO regression algorithm (Fig. 1C), while the RF algorithm identified four diagnostic genes (Fig. 1D). These four genes were identified as robust diagnostic biomarkers by using a Venn diagram (Fig. 1E). A ROC analysis of the GSE12285 dataset found that METTL3, RBMX, YTHDC1, and YTHDF2 were effective in discriminating between first-trimester and third-trimester samples (Fig. 1F).
GSVA and GSEA analysis and experimental validation
METTL3, an m6A-modified methylesterase, was found to exhibit significant differences in GSEA analysis across different expression groups, specifically in the calcium signaling pathway, intestinal immune network for IgA production, and N glycan biosynthesis (Fig. 1G). Further analysis through GSVA revealed that the high-expression group of METTL3 was highly enriched in primary immunodeficiency, lysine degradation, and intestinal immune network for IgA production, while the low-expression group was mainly enriched in maturity-onset diabetes of the young, proteasome, and regulation of autophagy (Fig. 1H).
To verify the importance of METTL3 in placental development, the expression of METTL3 in placentas from the first-trimester group and third-trimester group was examined. It was discovered that the first-trimester placentas exhibited significantly lower expression of METTL3 compared to the third-trimester group (Fig. 1I, J).
Subtype analysis of m6A regulators
In order to delve deeper into the role of m6A regulators in placental development, the consensus clustering method was used. The third-trimester samples were classified into two distinct subgroups, namely m6A clusters C1 and C2, using a k value of 2 as illustrated in Fig. 2A. The t-SNE results underscored a pronounced differentiation between these two m6A clusters, as depicted in Fig. 2B. Subsequently, we employed single-sample gene set enrichment analysis (ssGSEA) to quantitatively assess variations in infiltrating immune cells between the two m6A clusters and establish correlations between the expression of DEm6AR and immune cells (Fig. 2C). Significant discrepancies in various immune cell types were observed, including Activated B cell, Activated CD4 T cell, CD56 bright natural killer cell, Eosinophil, Gamma delta T cell, Immature dendritic cell, Macrophage, Mast cell, Monocyte, Type 2 T helper cell, Central memory CD8 T cell, and Effector memory CD8 T cell, all of which exhibited distinct distributions between the two clusters. Notably, a negative correlation was identified between METTL3 expression and a range of diverse immune cell types. Moreover, we meticulously examined variations in immune cells with either high or low METTL3 expression (Fig. 2D), revealing a substantial enrichment of immune cells characterized by lower levels of METTL3 expression. This observation strongly suggests a close association between reduced METTL3 expression and immune responses.
Furthermore, our investigation brought to light the upregulation of METTL3, WTAP, YTHDC1, HNRNPA2B1, and HNRNPC in m6A cluster C1, while RBMX, IGFBP2, CBLL1, and RBM15 exhibited higher expression levels in m6A cluster C2 (Fig. 2E). In our pursuit to comprehend the potential biological implications of the m6A-related cluster, we identified differentially expressed genes (DEGs) within this cluster by comparing m6A cluster C1 with m6A cluster C2 (Fig. 2F). Importantly, GSEA analysis revealed a significant enrichment of immune-related terms within these DEGs (Fig. 2G).
Construction of the m6A gene signature
We categorized the third-trimester samples into two distinct gene clusters, designated as gene clusters C1 and C2, based on the differential expression of genes associated with the m6A cluster. This partitioning strategy allowed for a targeted exploration of specific regulatory mechanisms (Fig. 3A). In the heatmap, a clear pattern emerged: gene cluster C1 encompassed 237 up-regulated genes and 462 down-regulated genes (Fig. 3B). Moreover, these gene clusters exhibited significant variations in the expression levels of DEm6AR (Fig. 3C).
Furthermore, the utilization of single-sample gene set enrichment analysis (ssGSEA) illuminated discernible discrepancies in the distribution of immune cells between the two gene clusters (Fig. 3D).
Correlations between m6A subgroups
Principal component analysis (PCA) was employed to calculate the m6A score. Notably, there were significant differences in the m6A score between the m6A clusters and gene clusters (Fig. 3E, F), with both m6A cluster C2 and gene cluster C2 displaying elevated m6A scores. This observation suggests a potential connection between the m6A score and immune activation. This trend was consistently mirrored in the levels of infiltration by various immune cell types, including Activated B cells, CD56 bright natural killer cells, Eosinophils, Gamma delta T cells, Immature dendritic cells, Macrophages, Type 2 T helper cells, Central memory CD8 T cells, and Effector memory CD8 T cells, in both m6A cluster C2 and gene cluster C2. To provide a visual representation, the distribution of the two m6A clusters, two gene clusters, and two m6A scores was depicted using a Sankey diagram (Fig. 3G).
WGCNA co-expression analysis
Firstly, WGCNA analysis of clinical trait correlations was performed to screen out the top 25% genes with the largest fluctuation for further WGCNA analysis, with outliers removed. Figure 4A shows that when power = 8, scale independence reached 0.9 and mean concordance was high. To detect outliers, we constructed trees using the module’s feature values, and then trees belonging to the same branch with very close distances are merged with the intercept value set to 0.5 (Fig. 4B). Figure 4C shows co-expression modules built by merging similar modules. Correlation analysis was performed to identify modules associated with specific traits based on the eigenvalues of the samples in each module and the characteristics of the samples, with genes represented in red being highly positively correlated in mature placentas. (Fig. 4D).
We followed the same approach as our WGCNA analysis of clinical trait correlations to analyze the correlations after typing, and we excluded outlier samples. Using a power = 7, we achieved scale independence of 0.9 and identified modules by color (Fig. 4E). We then discovered post-typing features and merged similar modules (Fig. 4F, G), which resulted in the identification of genes highly positively correlated in m6A cluster C1, represented by the dark red color (Fig. 4H).
Identification and functional enrichment analysis of METTL3-related genes
Through the intersection of clinical traits and gene modules with specific common traits, a total of 94 genes were pinpointed (Fig. 4I). Subsequently, by conducting correlation analysis, 42 genes closely associated with METTL3 was identified (Fig. 4J). Following this, functional enrichment analysis was performed (Fig. 4K), revealing that genes linked to METTL3 exhibited functional enrichment in processes including positive regulation of chemokine secretion, negative regulation of cytoskeleton organization, regulation of type 2 immune response, and type 2 immune response.
Identification of m6A-targeted immune genes and PPI analysis
We identified DEGs between the first-trimester and third-trimester groups (Fig. 5A). Subsequently, through the integration of genes identified using a three-step process, we successfully identified m6A modification-related immune genes (Fig. 5B). In order to examine the interactions between these immune genes related to m6A modification, we conducted a PPI analysis (Fig. 5C). Degrees, DMNC, MCC, and MNC were used in our analysis to identify genes. From their intersecting genes, we identified 5 hub genes (Fig. 5D). Hub genes are functionally enriched in the MAPK signaling pathway, focal adhesion, rap1 signaling pathway and ras signaling pathway (Fig. 5E).
Immune cell infiltration analysis
We used ssGSEA to quantify immune infiltration and obtain the enrichment levels of 27 immune cells and immune-related functions in the samples (Fig. 6A). The correlation between the 27 immune cells is shown in Fig. 6B. In addition, the correlation between these immune cells and hub genes was analyzed (Fig. 6C).
Validation of the hub genes
Then, five hub genes in the dataset GSE12588 were verified, and all these five genes were found to be upregulated in the dataset (Fig. 7A). The relevant pathways from the five gene pathway enrichment were then called up for visual analysis (Fig. 7B). And to further verify their efficacy, we validated the 5 hub genes in GSE12558 (Fig. 7C) and GSE9986 (Fig. 7D). ROC results indicated that all hub genes can discriminate between the first-trimester and third-trimester samples effectively in both datasets. We found that METLL3 could target all these genes, and among these gene-related pathways, the focal adhesion pathway could target all four genes at the same time. What’s more, the FAK signaling pathway may have an impact on the early development and function of the IVF-ET placental villi.
Moreover, our in vivo animal experiments have found that the mRNA expression of all these 5 genes in the first-trimester placentas was significantly lower than that in the third-trimester placentas, which is consistent with our results above (Fig. 7E).
Experimental validation of the Pregnant Rat Models of Fear Stress
Our sequencing study found that the expression of METTL3 was significantly downregulated in fear stress group (Fig. 8A). Thus, we utilized the pregnant rat model of fear stress to investigate whether METTL3 affects placental vascular development by targeting the five immune-related genes.
Compared to the control group, the third-trimester rats with fear stress exhibited heightened anxiety, as evidenced by a significant preference for the periphery of the arena in the open field test (Fig. 8B, C), as well as an increased duration spent and entries in the closed arm in the elevated plus maze test (Fig. 8D). These findings indicate the successful establishment of the fear stress model. As shown in Fig. 8E, the size of placentas was smaller in the fear stress group than in the control group. Additionally, the weight of the offspring and placentas was lower in the fear stress group (Fig. 9A). The mRNA expression of five hub genes in the fear stress group was significantly down-regulated compared to the control group (Fig. 9B). Transmission electron microscopy results showed that in the control group, placental vascular endothelial cells were tightly connected, with abundant microvilli on the cell surface, and intact nuclear membranes. In contrast, the fear stress group exhibited evident signs of vascular damage in the placentas, with increased proliferation of endothelial cells protruding into the lumen, and a reduction in surface microvilli (Fig. 9C).
{kind=link}