Ethics statement and clinical tissue samples
Our study was approved by the Independent Ethical Committee of the First Affiliated Hospital of Nanchang University and complies with the Declaration of Helsinki. Written informed consent was received from all patients. Eight primary GC tissues and their corresponding normal gastric tissues were obtained from the Department of Surgery at the First Affiliated Hospital of Nanchang University. Upon resection, the fresh tissue samples were immediately frozen in liquid nitrogen and stored at -80 °C refrigerator. The clinicopathological characteristics of GC patients are summarized in Additional file 1: Table S1. A total of 161 paraffin-embedded gastric cancer samples were included in this study. All GC patients were treatment-naïve before surgery. The clinicopathological features of the GC patients were confirmed by two experienced pathologists at the First Affiliated Hospital of Nanchang University between 2012 and 2016. The clinical and pathological grade was conducted according to the eighth edition of the classification system of the American Joint Committee on Cancer (AJCC).
Cell Lines And Cell Culture
The human GC cell lines AGS, BGC823, MGC803, HGC-27, SGC7901 and MKN45 and the immortalized gastric epithelial cell line GES-1 were purchased from the Shanghai Institute of Cell Biology, China Academy of Sciences. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, Logan, UT, USA) with 10%-15% fetal bovine serum (FBS; HyClone, USA) at 37 °C in an atmosphere containing 5% CO2. Cells were harvested at the indicated times posttransfection for future experiments.
Vectors, Lentiviral Infection And Transfection
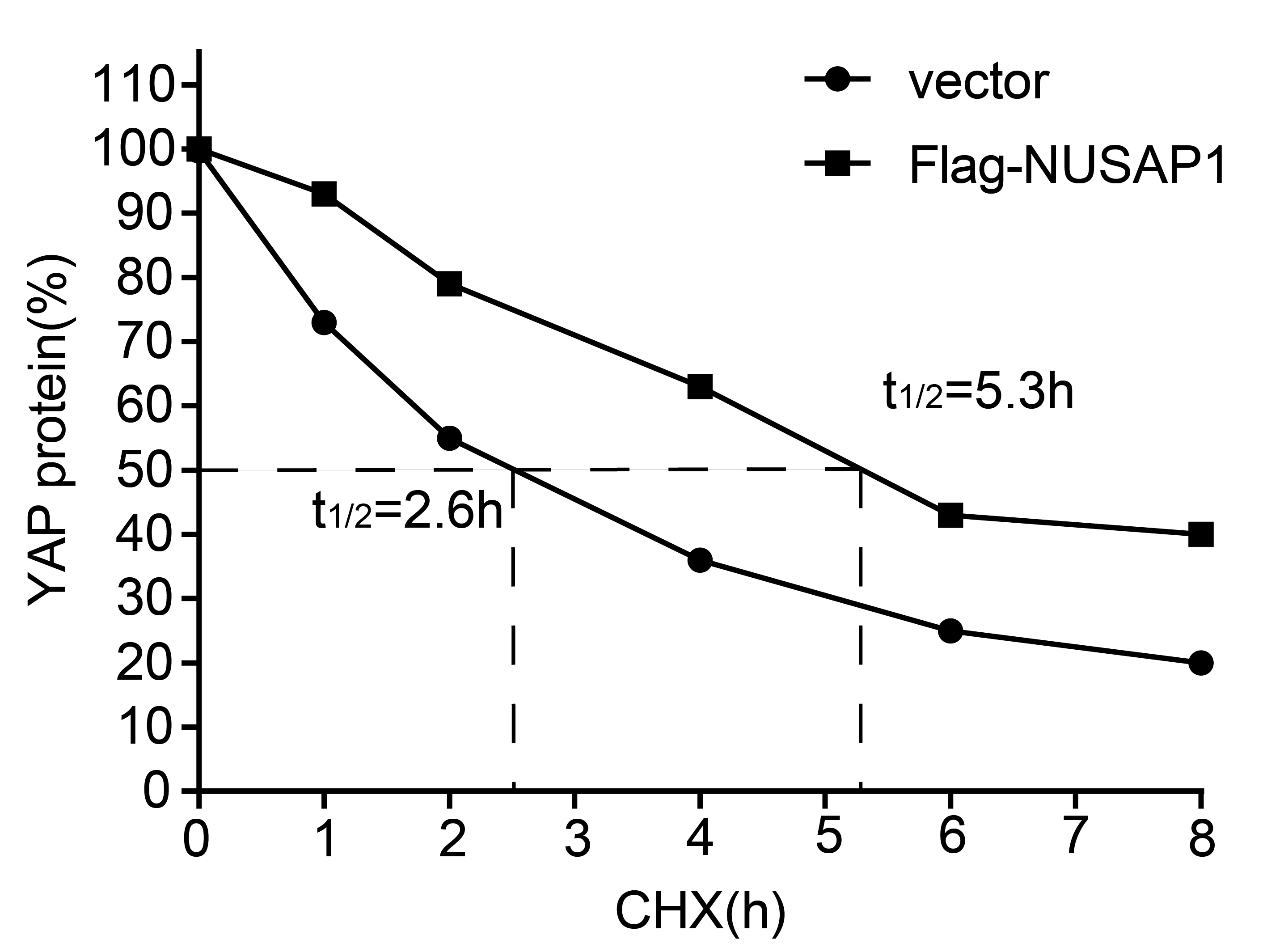
Flag-NUSAP1 plasmid was generated by subcloning the PCR-amplified human NUSAP1 coding sequence into the 2 × pcDNA3.1 vector at the NheI and HindIII sites. NUSAP1-targeting shRNAs and YAP-targeting siRNA oligonucleotides were designed and synthesized by GenePharma. The sequences of shRNAs and siRNAs used in our study were provided in Additional file 2: Table S2. Cells were seeded on the plate the day before transfection, and transfected with indicated plasmids, shRNAs and siRNAs using TurboFect transfection reagent (Thermo Scientific, R0532, USA). Lentivirus production and infection were conducted as described previously[27]. Stable GC cell lines transfected with scramble shRNA or NUSAP1 shRNAs were selected for two weeks and incubated with 2 µg/mL puromycin upon infection.
Western Blotting Analysis
Western blotting analysis was conducted using NUSAP1 (1:1000; #12024-1-AP; Proteintech, Rosemont, IL, USA), YAP (1:1500; #14074; Cell Signaling Technology, Danvers, USA), LATS1 (1:1500; #3477; Cell Signaling Technology, Danvers, USA), LATS2 (1:2000; #5888; Cell Signaling Technology, Danvers, USA), CTGF (1:2000; #86641; Cell Signaling Technology, Danvers, USA), CYR61 (1:2000; #14479; Cell Signaling Technology, Danvers, USA), anti-HA (1:2500; #ab9110; Abcam, Cambridge, USA), anti-Flag (1:2500; #A2220; Sigma-Aldrich, USA), β-actin (1:3000; #AF7018; Affinity, Jiangsu, China) and GAPDH (1:3000; #A2220; Affinity, Jiangsu, China). Human GC samples originally obtained from the First Affiliated Hospital of Nanchang University were ground and lysed in lysis buffer before western blotting analysis. The western blotting experiments were performed as previously described[28]. GAPDH or β-Actin was used as an internal control, and all the protein bands were analyzed using ImageJ software.
Immunohistochemistry
Immunohistochemistry was performed in 161 clinical gastric cancer samples as reported previously[29]. The degree of immunostaining was assessed and scored by two pathologists in a blinded manner. The protein expression of NUSAP1 in gastric cancer specimens by determining the SI (the product of the staining intensity score and the proportion of positive cells), with possible scores of 0, 1, 2, 3, 4, 6, 8, 9, and 12. Samples with a score index ≥ 6 were considered to have high NUSAP1 expression, and those with a score index < 6 were considered as low NUSAP1 expression.
Rna Extraction And Quantitative Real-time Pcr
Total RNA was extracted from cells or xenograft tissues using TRIzol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol as previously described[30]. Total RNA (0.5 or 1.0 µg) was used as a template for reverse transcription using poly-(T)20 primers and M-MLV reverse transcriptase (Promega, Madison, USA). Quantitative RT-PCR (qRT-PCR) was performed using SYBR Green Mix following the manufacturer’s protocol (BioRad, Hercules, USA). The 2−∆∆Ct method was utilized to assess the relative mRNA expression of genes among groups. The primers sequences used are listed in Additional file 3: Table S3.
Cell Counting Kit-8 (cck-8) Assay
To assess cell growth ability, the CCK-8 assay (Beyotime, China) was utilized following the manufacturer’s instructions as previously described40. After transfection for 24 h, cells were inoculated in 96-well plates. Cell viability was measured by adding 10 µL CCK-8 reagent and incubated for 1 h, then the optical density was measured at the absorbance of samples at 450 nm in a microplate reader (SpectraMax M5e, USA) for 5 days. The data derived from triplicate samples are shown as the mean ± standard error of the mean (SEM).
Colony formation assay.
The colony formation assay was performed as previously described[31]. The indicated transfected HGC-27 and BGC823 cells were trypsinized and plated at equal numbers of cells into 6-well plates. Media were changed every 3–4 days until the colonies were visible. Colonies were fixed with 4% PFA (paraformaldehyde), and stained with 10% crystal violet at room temperature for 30 min. ImageJ software (Rawak Software, Stuttgart, Germany) was used for quantification of the colonies. These experiments were conducted more than three times.
Wound Healing Assay
Indicated HGC-27 and BGC823 cells were plated into 6-well plates at a concentration of 4.6 × 105 per well and serum-starved for 24 h until completely confluent on the second day. A sterile 200 µL pipette tip was used to draw straight lines to form a wound. The cells were carefully washed with phosphate-buffered saline (PBS) and cultured in serum-free medium. Images were taken at 0 h, 24 h or 36 h after scratching to evaluate wound closure. Each experiment was carried out for at least three times.
Transwell Assay
The transwell chambers were prepared with Matrigel gel, indicated HGC-27 or BGC823 cells were plated into the upper chambers as described previously[32]. After 72 h of incubation, cells passing through the chamber membranes were fixed with 4% PFA and stained with 10% crystal violet. These experiments were conducted using three biological replicates.
Immunofluorescence Staining
The indicated BGC823 cells were fixed in 4% PFA for 15 min and then treated with 0.5% Triton X-100 for 10 min. Non-specific bindings were blocked with 5% bovine serum albumin (BSA) for 30 min. The cells were incubated with the anti-Flag or anti-GFP antibodies at 4 °C overnight and incubated with the corresponding secondary antibody for 20 min. The 4’-6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei for 30 min. The cellular localization of NUSAP1 or YAP was detected using a confocal microscope (Nikon, ECLIPSE Ti2).
Co-Immunoprecipitation.
The Co-immunoprecipitation (Co-IP) assay was conducted utilizing indicated antibodies according to the figure legends as previously described40. Briefly, 1000 µg of total protein was incubated with anti-Flag beads (#M185-11 MBL, Tokyo, Japan) at 4 °C for 4 h. The beads were washed with lysis buffer for at least three times. Bound proteins were detected by Western blotting analysis with indicated antibodies described in the figure legends.
Animal Models
Six to seven-week-old female nude mice were purchased from the SLACCAS Experiment Animal Company (Shanghai, China). 5.6 × 106 indicated BGC823 cells were subcutaneously inoculated into the left axilla of each mouse. Tumor growth was monitored every 3 days with electronic digital calipers (Thermo Scientific) in two dimensions. Tumor volume was measured with the formula: tumor volume (mm3) = (length × width2)/2. After 28 days, the mice were sacrificed by euthanasia, and xenograft tumors were harvested and weighed. Total RNA was extracted from tumors via homogenization in TRIzol buffer and then subjected to qRT-PCR analysis.
Statistical analysis
The SPSS 20.0 software (Chicago, IL, USA) was utilized to perform the statistical analysis. Student’s two-tailed t-test or the chi-square test was used to determine the mean difference among groups. Survival curves were plotted using the Kaplan-Meier method and compared by the log-rank test. P < 0.05 was considered statistically significant. All the data are presented as the mean ± SEM.
{kind=link}