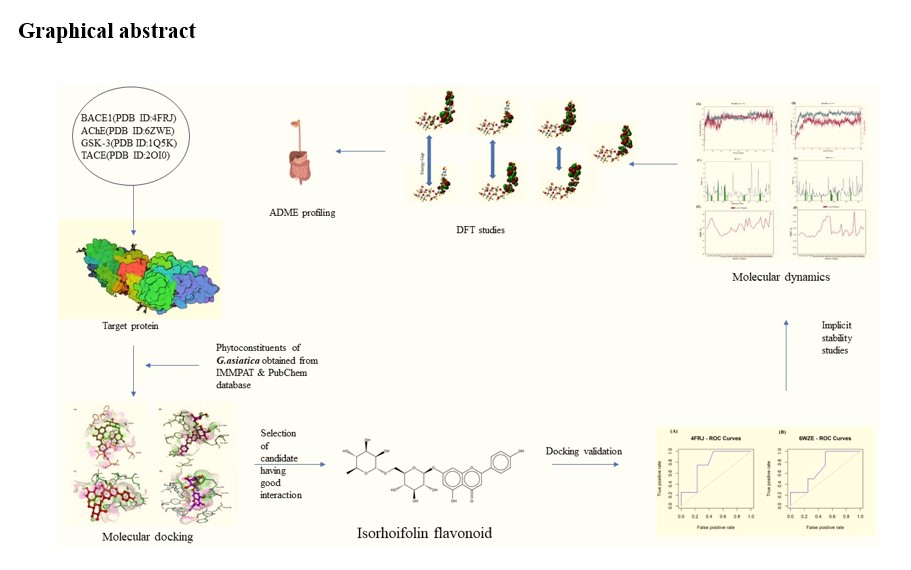
Molecular docking studies
Screening of phytochemicals by docking
Auto-dock software was used for the molecular docking studies [18]. Binding free energy and types of interactions are some of the essential parameters analyzed in the docking study. Table 1 represents each phytoconstituent's docking binding energy values with specific proteins. Among all constituents, isorhoifolin showed the least binding energy compared to the other phytochemicals in plant leaves.
Table 1
Binding energies of phytoconstituents with target protein receptors of AD.
| Sr No | Phytoconstituents | 6ZWE (AChE) [kcal/mol] | 4FRJ (BACE-1) [kcal/mol] | 1Q5K (GSK-3) [kcal/mol] | 2OI0 (TACE) [kcal/mol] |
| 1 | Cycloolivil | -7.6 | -7.4 | -7.2 | -7.5 |
| 2 | Spathulenol | -7.9 | -7.5 | -7.5 | -6.8 |
| 3 | Nicotiflorin | -9.1 | -9.9 | -9 | -8.5 |
| 4 | Gmelinol | -9.3 | -7.8 | -7.8 | -9.5 |
| 5 | Quercetagetin | -9.3 | -8.4 | -8.5 | -8.9 |
| 6 | Pinoresinol | -9.4 | -8.5 | -8.5 | -8 |
| 7 | Sakuranetin | -9.7 | -8.4 | -8.5 | -8.5 |
| 8 | Luteolin-7-glucoronide | -9.9 | -10.4 | -9.6 | -9.8 |
| 9 | Poulownin | -10.8 | -9.4 | -8.8 | -9.2 |
| 10 | Isorhoifolin | -10.9 | -10.2 | -9.7 | -9 |
Table 2
Interactions of target proteins with isorhoifolin
| S. No. | Target protein | Types of bond interaction | Amino acid residues |
| 1 | 4FRJ (BACE1) | Conventional hydrogen bond | ASN233, TRP76, GLN73, GLY230, ASP32, ARG235 |
| Alkyl | VAL332 |
| Vander Wall forces | THR232 |
| 2 | 6ZWE (AChE inhibitor) | Conventional hydrogen bond | PHE295, TYR341, TYR134 |
| Pi-Pi T shaped | PHE297, PHE338, TRP286 |
| 3 | 1Q5K (glycogen synthetase kinase GSK-3) | Conventional hydrogen bond | GLU185, VAL35 |
| Pi-Sigma | LEU189, CYS199, VAL70, ILE62 |
| Pi-Alkyl | LEU189 |
| 4 | 2OI0 (TNF-alpha converting enzyme) | Conventional hydrogen bond | GLN185, VAL135 |
| Pi-Sigma | VAL70, ILE62 |
| Pi-alkyl | LEU188, CYS199, VAL62 |
In the BACE1-isorhoifolin complex, the formation of conventional hydrogen bonds with oxygen and hydrogen of the phenyl ring with amino acid ASN233 and Vander Waals bonds with THR232, respectively, makes stable bond formation of the complex. Other than phenyl ring cyclohexyl and phenolic hydrogen with oxygen creating a hydrogen bond with amino acid residues of TRP76, ASP32 enhances the complex's stability. Alkyl bond formation between VAL332 of the amino acid residue and hydrogen of the oxy-cyclohexyl ring shows the saturation nature of the ligand isorhoifolin, which helps the ligand for the assembly of the complex (Fig. 2).
For the AChE inhibitor-isorhoifolin complex, there was a various nature of bond formation that helped the ligand form the anchored composite. Conventional and Vanderwalls bonds formed between isorhoifolin and amino acid residues TYR124 and GLY121, respectively (Fig. 2). Apart from these interactions, ketonic oxygen and phenolic hydrogen of isorhoifolin interact with the target protein's PHE295 and TYR341 amino acid residues, creating a hydrogen bond. TRP286 and PHE297 residues of AChE target protein (6ZWE) had pi-pi stacked T-shaped bond attraction with the phenolic ring and ketonic oxycyclohexyl ring of isorhoifolin, resulting in sturdy attributes of complex (Table 2).
The GSK3-isorhoifolin conglomerate had profuse bond intricacy, where there was an unfavorable donor-donor bump between cyclohexyl and phenolic hydrogen of isorhoifolin with CYS183 and TYR138 amino acid residues of target protein GSK-3. Besides these conventional bonds between oxy cyclohexyl and phenolic hydrogen with VAL135, GLN185 amino acid residues of the target protein compensate for the bump of donor-donor. Pi-pi sigma interactions are seen between amino acid residues VAL70 and ILE62 with quinolic and phenolic hydrogen of ligand (Fig. 2). The last pi-sigma nature is also seen in the complex where LEU188 and CYS199 amino acid residues are fused with phenolic and quinolinic hydrogen of ligand.
TACE target protein (2OI0) with isorhoifolin ligand network conventional hydrogen, van der Walls, amide-pie stacked, pi-alkyl, and one bump glimpsed. Amino acid residues for conventional and Vander walls hydrogen bond involved are ASW447, GLU406, and PRO437 with oxygen, hydrogen of phenol and quinol, and cyclohexyl carbon, respectively. Saturating nature bonds like amide-pi stacked and pi-alkyl also contribute to the complex's higher binding energy than the other phytoconstituents of plants. TYR433 of residues with phenyl ring LEU401, VAL434 residues with phenolic hydrogen, and VAL402, ALA439, and LEU348 of residues with quinolinic hydrogen of isorhoifolin had the solid interaction (Fig. 2).
The above synergy of isorhoifolin with the target protein is responsible for its high binding energy, contrary to the other phytochemicals of the plant. Hence, we used isorhoifolin phytoconstituent for further molecular dynamics and DFT calculation. The most stable complex for the MD simulation analysis was selected based on the binding energy of the complex. Table 1 shows that BACE1-isorhoifolin and AChE inhibitor-isorhoifolin had the highest binding energy, -10.2 and − 10.9, respectively. These two complexes were further analyzed for MD studies.
Docking validation
The perceptible illustration of the correlation between the contender of test precision and vulnerability was computed throughout the ROC curves. The ROC curve generation was initiated by delineating the rate of authentic positives proportionate to the percentage of false positives commensurate to the percentage of true negatives. The outlined ROC curve design was availed to substantiate the chosen phytoconstituents for molecular docking studies so that designated compounds should be from functional ligands rather than idle ligands (decoys). Furthermore, the delineated pattern differentiated the active ligands from the best-in-class phytoconstituents in the chosen database. The values 0.7788462 and 0.708333 are discernible areas under the curve, and an enrichment factor was discovered in the top 1% at 3.25 and 3.05 as well grounded (Fig. 3). The ROC curve is the correlation between responsiveness and specificity. It corresponds to the presentations of actual positive and false positive fractions on the y-axis and x-axis. The values 0.7788462 and 0.708333 are good areas under the ROC, representing that the docking gimmick played a significantly specific and accurate role in docking with target receptor proteins 4FRJ and 6ZWE, respectively, with isorhoifolin.
Molecular dynamics studies
A RESPA integrator was employed to amalgamate all the frameworks of the dynamic environment of interlinkage and component firmness. Docking concordats are usually expeditious and coarse-grained; nevertheless, docking scantiness protein pliability can mediate with the reliability of the concomitant target protein-ligand complexes. Therefore, a detailed molecular dynamics simulation methodology might ameliorate a preferable harmonizing with docking [19]. Substantially, MD is accustomed to gauging macromolecular and contingent variables based on conventional mechanics and by using Newton's analogy of motion to reckon the speed and locus of each atom of the contemplated organization. That means MD accomplishes a more rigorous vindication than docking studies. The study helps in the solvation investigation of molecules along with characteristics of the target protein and protein-ligand interlinkage, perhaps investigated through MD [20]. This study selected the most favoured constituents for further dynamics study based on binding energy and numbers with the type of interaction through molecular docking study. It was observed that isorhoifolin was the compound that had the highest binding energy and a good number of interactions as compared to other phytoconstituents of G. asiatica. Utilizing the best complexes and docking conformations found from Auto Dock, they were examined for plausible atomic characteristics within the solvent system [21]. For that purpose, complexes of BACE1-isorhoifilon and AChE inhibitor-isorhoifolin were further selected for dynamics study. Interchange allying protein along with ligand is conceivably elucidated by PL and LP proximity bar representations (Fig. 4A) for ligand fleck (Fig. 4B). The diagram manifests assorted peculiar interactions of the ligature in the company of amino acid residues of the target protein, additionally designated as hydrogen, hydrophobic, ionic bonds, and water aqueducts [22]. Two target protein-ligand complexes, BACE1-isorhoifolin and AChE inhibitor-isorhoifolin, were deliberated through MD simulations to inaugurate the steadiness of the complex. The comprehensive perusal of trajectories acquired from the MD run was scrutinized for target proteins BACE1, AChE inhibitor, and ligand isorhoifolin RMSD, RMSF, torsion portrayal, and hydrogen bonding [23].
The Fig. 4 bar illustration showed hydrogen bonding with amino acid residues GLY 32, TRP 36, PRE108, ARG235 for BACE1-isorhoifolin complex whereas for AChE inhibitor-isorhoifolin seemed to be THR95, GLU292, SER293. Ionic bonding with amino acid residues ASP32 & 298 for BACE1-isorhoifolin and AChE inhibitor-isorhoifolin complex was found to be with ALA204, ASN283. For hydrophobic interactions, residues involved were THR71 and ILE118 of the BACE1-isorhoifolin complex, whereas, in complex 6ZWE-isorhoifolin, these interactions are with amino acid residues of MET85, GLY122, GLU285, TYR337 & VAL340.
Protein RMSD outlines the gesticulation of divergent ligand atoms contemporary in the active site of the target protein receptor and anticipates the structural contour of the protein throughout MD. The consummate aberration in RMSD for most globose proteins is contemplated in the ambit of 1–4Å. RMSD of 4FRJ and 6ZWE come across protein mainstay RMSD of 1.8 Å & 2.8 Å, and for ligand, it was shown deviation of 1.5 Å & 1.8 Å.
In the ligand RMSD plot context, the Lig-fit port indicates a valued subordinate to the protein RMSD, prudent that the ligand is conscientiously disseminated to the commencing binding site and contrariwise. The orientation also proclaims that the ligand is well resided inside the binding site Fig. 5 (A) & (B). RMSF succor in prognosticating the structural rectitude of the target receptor protein-ligand complex constituted by RMSF nomograms P-RMSF and L-RMSF. The P-RMSF delineates provincial transpose in the protein chain, forasmuch as L-RMSF Fig. 5 (C), (D), (E), and (F) chronicle commute in the orientation of ligand atoms. In the bargain, protein RMSF (P-RMSF) is acclimated to appraise the obligatory steadiness of the protein by distinguishing the fluctuation in the vertexes of the plot throughout the simulation. The standard P-RMSF was correspondingly considered 1.8 Å & 2.4Å for BACE1-Isorhoifolin & 6ZWE-Isorhoifolin complexes. The ligand RMSF exhibit for BACE1-isorhoifolin AChE inhibitor-isorhoifolin was discovered to be 6 Å and 3 Å, respectively.
Ligand contortion is a requisite parameter to scrutinize the energy dynamic obligation of ligand conclusive free energy, with the numerical signification of the malleable. The contour exchanges that each rotatable bond (RB) afforded by ligand molecule that encounters all over its MD miniature could be outlined from its ligand torsion silhouette. In Fig. 6, assorted rotatable bonds inside the ligand structure are colour-enciphered diversely and portrayed with a dial and bar plot intimated in colour to communicate to the corresponding bond. The phase of the bar plot constitutes the prospect of torsions as a corollary of angle, and the dial plot exhibits the torsional angle as it synchronizes and lengthens cochlearly toward the edge throughout the simulation. isorhoifolin expressed the occupation of four rotatable bonds in the middle of carbon atoms of phenyl aliphatic ring for a protein 4FRJ & 6ZWE complex.
Assets like hydrogen bonds necessitate consideration for developing novel advanced drugs that cause hydrogen bonding to have a consequential notable influence on drug exactitude, absorption, metabolism, etc., due to association in the ligand binding. Subordinates of hydrogen bonding are glimpsed in the protein-ligand binding, which is the backbone donor and acceptor as well as side chain doner acceptor complex of protein 4FRJ & 6ZWE expressed interaction of hydrogen bond with residues of amino acid and for complexes. Scrutinizing the Vander walls radii of 1.4 Å, SASA was computed as median SASA for ligand against target receptor protein (4FRJ & 6ZWE) diminished after binding with complex (Fig. 6).
Succinctness of interaction abides by rGyr; underneath values of rGyr corresponds to higher steadiness and vice versa. The plot of rGyr vs. time of complex BACE1-isorhoifolin & AChE inhibitor-isorhoifolin interposed. Median rGyr for complex BACE1-isorhoifolin and AChE inhibitor-isorhoifolin appeared to be 22 Ų 23.2 Ų, respectively, throughout MD simulations run of 200 ns (Fig. 7).
Density functional theory (DFT) studies
It was accomplished to optimize the isorhoifolin structure (Fig. 8A). The discrete atomic bond parameters, such as bond length angle, have been established. The C = O bond length obtained was 1.5 Å, whereas, for the C-H bond, it was 2 Å. To a more significant extent, sites of electrophile nucleophiles were investigated using MEP surface inspection (Fig. 10A). The colour of MEP surfaces (red, yellow, blue, orange, green) indicates charge distribution [24]. The range of charge distribution was − 5.883e-2 to + 5.883e-2. In Fig. 10 (B), the coloured region indicates bulk negative electrostatic potential, whereas the coloured region stipulated the most positive electrostatic potential. A green colour indicates the neutral electrostatic potential. Oxygen atoms show high electromagnetic potential on MEP surfaces due to their electrophilic reactive nature, whereas carbon atoms show nucleophilic nature [25].
HOMO (Highest Occupied Molecular Orbital) & LUMO (Lowest Unoccupied Molecular Orbital) are merely FMO (Frontier Molecular Orbitals) which are beneficial to recognize numerous chemicals, thermal, and electronic assets of molecules. Energy difference allying HOMO & LUMO of molecules shown by the density of state (DOS), furthermore reactivity of global and local variables was also scrutinizing, for instance, energy aperture, electron energy, chemical solidity as well as downiness, electrophilicity lead intimation, ionization potential [26].
The energy difference between FMO, i.e., and HOMO-LUMO, is -0.162, showing ISF is more reactive. In the structure of ISF, the aromatic ring region appeared to be LUMO surfaces, whereas the aliphatic regions appeared as HOMO regions (Fig. 9). Phytoconstituents expressed equilibrated LUMO energy that fact stipulates ISF exhibit biological solid activity. Those, as mentioned earlier, might be expected because of the existence of a ketonic group. Carbonyl operates as an electron revulsion class. In the report of Koopman's theorem, global local parameters such as LUMO energy characterize a reminiscent part of pharmacological action. Outright hardness with a softness framework can be accustomed to prognosticate phytocompound isorhoifolin excitability and steadiness.
Mulliken occupant's charges, bond length, bond order exploration
Gaussian 6.0.16 software assessed the Mulliken charges, bond length, bond order, etc. These parameters implicate the inclination of ISF phytoconstituents towards the absorption of electrons. The positive charge is exhibited by H and C atoms, whereas oxygen atoms show the negative charge (Fig. 10). Dual descriptors were used to calculate the Fukai function, identifying the difference between electrophile nucleophiles. Higher electrophilic sites stipulate the reactivity of ISF toward biological activity as well as the system.
The following generation of HOMO (-0.2267 ev) and LUMO (-0.06358 ev) and elevated energy gap (-0.162 ev) represent that a high-rise charge transmission takes place with isorhoifolin & creates a steady interaction with target protein concerning to the intensification of the bioactivity (Fig. 10B). The value − 0.162 ev stipulates that the molecule has robust inhibition effectiveness due to energy requirement in removing an electron from hindmost molecule inhibitors acquire free electron moreover donate an electron to unoccupied orbital, making them supplemental electron-rich and thus offering supercilious inhibition efficiency. The results showed that isorhoifolin is occupied with electrons since the LUMO value represents a molecule caliber to receive free electrons, supply free electrons, and provide excellent inhibition accomplishment.
ADMET Attributes
The ADMET characteristics of a composite are a predominant feature of its unsympathetic potency. The SwissADME server bestows an adaptable, accessible web frontier to speculate copious ADME parameters [27]. Assimilation of the drugs becomes a restraining facet for their bioavailability and retaliation. A drug is deliberated competently if absorbed in the human intestine exceeding 70% [28]. There is an adaptation of philosophy in the pharmaceutical industry: If a drug fails early, it will fail competitively, which asserts that vigor action, i.e., potency, is not at most property concernment [29]. This emanated in incorporating in-vitro and in silico, ADMET parameters at the initial and development stages. Such an approach has the intense possibility of astonishing declination, which is observed in the ADME point at issue seen with the termination of projects [19]. The phytoconstituents of plant G. asiatica were analyzed for ADME and toxicity properties the pkcsm server was used for ADME profiling of phytoconstituents with toxicity studies (Table 3).
Computational analysis for drug likeliness with infinitesimal properties sieving
Roughly, drugs are defined as a class of chemically active entities which are having potency, affinity, and intestinal therapeutic efficacy with the target; other than these characteristic properties, they should also fulfill indubitable criteria like bioavailability at the same time devoid of toxicities acute/chronic, any type of carcinogenicity, mutagenicity, genotoxicity, etc. Hence, that objective ADMET framework was enumerated and examined for harmony with their standard compass. A phytoconstituent with high CaCo2 permeability is 0.392, easily absorbed if the Papp coefficient is more than 8 x 10− 6. Besides CaCo2 permeability, if the absorption of the drug is less than 30%, it is a poor candidate for formulation purposes. All the phytoconstituents were anticipated to have sound absorption at the intestine for skin permeability. Log kp values are considered; if it is less than − 2.5, it will have low permeability related to skin [9].
Table 3
ADMET profiling of isorhoifolin.
| S. No. | ADMET attributes | Property name (unit) | Speculated values |
| 1 | Absorption | Skin permeability (log Kp) | -2.735 |
| Water solubility (log mol/L) | -3.262 |
| Caco2 permeability (LOG Papp in 10-6cm/s) | 0.392 |
| P-glycoprotein substrate | Yes |
| P-glycoprotein I inhibitor | No |
| P-glycoprotein II inhibitor | No |
| Intestinal absorption[human](%Absorbed) | 30.186 |
| 2 | Distribution | Fraction unbound [human](Fu) | 0.089 |
| BBB permeability (log BB) | -1.845 |
| CNS permeability (log PS) | -5.145 |
| VDss[human] (log L/kg) | 0.269 |
| 3 | Metabolism | CYP2D6 substrate | No |
| CYP1A2 inhibitor | No |
| CYP3A4 substrate | No |
| | CYP2C19 inhibitor | No |
| CYP2D6 inhibitor | No |
| CYP2C9 inhibitor | No |
| 4 | Excretion | Renal OCT2 substrate | No |
| Total Clearance (log ml/min/kg) | 0.451 |
| 5 | Toxicity | Hepatotoxicity | No |
| Skin sensitization | No |
| T. Pyriformis toxicity (log ug/L) | 0.285 |
| Minnow toxicity (log mM) | 5.005 |
| Oral rat acute toxicity [LD50] (mol/kg) | 3.903 |
| hERG_ I inhibition | No |
| hERG_II inhibition | Yes |
{kind=link}