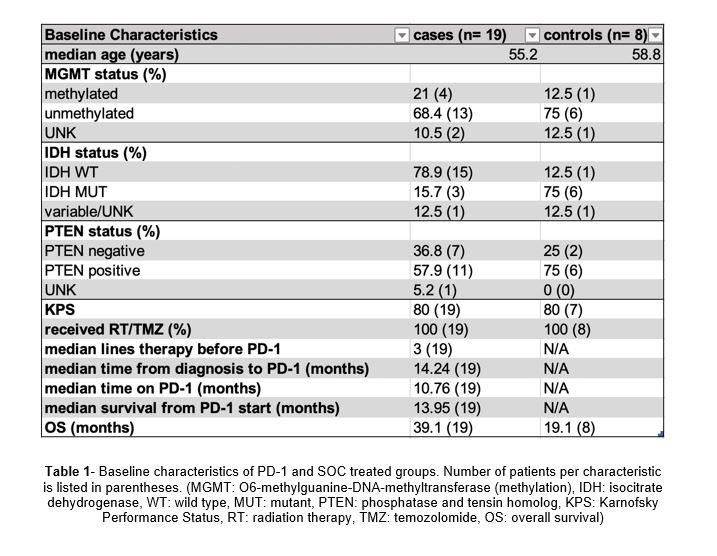
Glioblastoma multiforme (GBM) is the most aggressive primary brain tumor (glioma) in adults (WHO Grade IV) and accounts for more than half of all brain tumors [1]. Even after surgery, chemotherapy, and radiation therapy, tumors often recur and survival remains poor at fewer than 15 months after diagnosis [2]. Common pathological biomarkers include methylation (MGMT) status, isocitrate dehydrogenase (IDH) mutation, epidermal growth factor receptor (EGFR) amplification, and phosphatase and tensing homolog (PTEN), all of which have prognostic relevance or may be predictors of response to specific therapy. PTEN is a tumor suppressor gene that plays an integral role in cell proliferation regulation, adhesion and invasion, apoptosis, and DNA damage repair. PTEN loss in melanoma tumor cells increases expression of immunosuppressive cytokines, which leads to decreased T cell infiltration in tumors, and inhibition of autophagy, which regulates T cell-mediated cell death [3]. Loss of PTEN expression begins early in glioma development with mutations occurring in between 5% and 40% of cases [4]. Zhao et al. demonstrate in their cohort of GBM patients treated with immunotherapy that non-responders had tumors enriched in PTEN mutations. RNA-sequencing has also indicated that the PTEN mutation may induce an immunosuppressive microenvironment and lead to shorter overall survival. As a result, PTEN status has become a valuable biomarker in the investigation of the tumor microenvironment in GBM.
Standard of care (SOC) therapy typically includes maximal surgical resection followed by radiation therapy and concomitant and adjuvant temozolomide. Better treatment strategies are urgently needed and immunotherapies such as programmed cell death protein-1 (PD-1) inhibitors are in clinical trials for the treatment of GBM. Immune checkpoint inhibitors of PD-1 have revolutionized the treatment of other cancer types, such as lung cancer and melanoma, by blocking PD-1 (on effector T cells) which interacts with PD-L1 on the surface of cancer cells to suppress immune function. Programmed death-ligand 1 (PD-L1) testing helps identify patients who may benefit from anti-PD-1 therapy in many tumor types. In GBM specifically, PD-L1 is not a predictive marker for response to PD-1 or PDL-1 checkpoint inhibitors, due primarily to the variance in expression and subcellular distribution of PD-L1 in glioma cells [5]. A large phase 3 study using the PD-1 inhibitor nivolumab in recurrent glioblastoma (Checkpoint 148 from BMS) showed an 8% overall response rate in GBM but no predictive biomarker of response [6]. Finding a biomarker that can predict response to PD-1 inhibitors would be extremely helpful in GBM as this would allow for the selection of patients most likely to benefit from such therapy.
The lower efficacy of PD-1 inhibitors in GBM may be due to microenvironment immunosuppression and low tumor mutational burden (TMB). The higher the number of mutations per coding area of a tumor genome yields a more stimulated immune response and more favorable tumor response to immunotherapy. Hodges et al. discovered that high TMB was found in only 3.5% of GBM patients and that both high and moderate TMB GBMs did not have an enriched influx of CD8+ T cells, PD-1+ T-cells, or tumor-expressed PD-L1 cells [7]. They also demonstrated that the subset of GBMs that should have a better response to immune checkpoint inhibitors based on TMB still lacked the typical biomarkers of response, further suggesting the limited significance of PD-1 and PD-L1 testing in GBM. Nevertheless, finding a more specific and accurate testing of these biomarkers could be beneficial in treating certain GBMs due to the severe lack of proven treatment options.
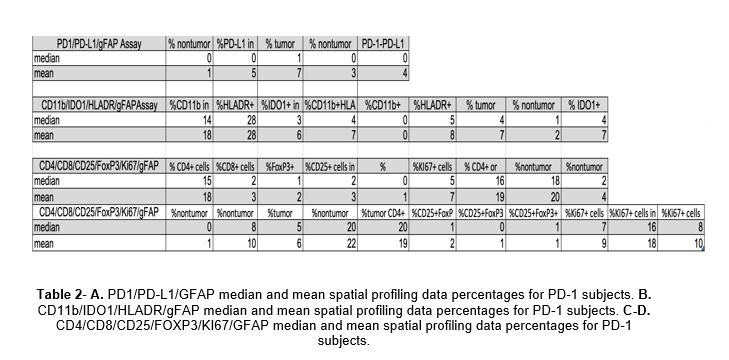
Multiplex immunostaining panel technology has explored novel biomarkers in melanoma and identified two which have prognostic significance in overall survival, namely PD-1/PD-L1 interaction score, which measures density PD-1-positive cells in proximity to PD-L1-positive cells in contrast to measuring PD-L1 alone, and colocalization of human leukocyte antigen-DR (HLA-DR, D= aspartic acid, R= arginine) and indoleamine-2,3-dioxygenase-1 (IDO-1), which are upregulated by interferon gamma (IFNy) in tumors rich with immune cells [8]. Our primary hypothesis was that although response to PD-1 immunotherapy in GBM is low, multiplex immunostaining panel technology is feasible on GBM tumor tissue and will allow for detailed analysis of tumor microenvironment cells and their interaction with one another. Our secondary hypothesis was that PD-1 immunotherapy based on PD-1/PD-L1 interaction score and IDO/HLA-DR expression can predict overall survival (OS) and survival from initiation of PD-1 therapy.
HLA-DR is an MHC class II cell surface receptor that presents peptide antigens to the immune system to mediate T cell response [9]. IDO-1 is a tryptophan-metabolizing enzyme that regulates immunologic tolerance and whose expression varies across tumor types [10]. IFNy is a cytokine essential to innate and adaptive immunity, particularly in the activation of macrophages and inducer of class II MHC molecule expression [11]; it acts to limit tumor growth and induces IDO and HLA-DR expression on tumor cells. Johnson et al. demonstrated that high PD-1/PD-L1 interaction and high IDO-1/HLA-DR were statistically significant different between responders and non-responders; however, coexpression of both biomarkers had the best response to anti-PD-1 therapy and was correlated with the best survival outcome [8]. Unfortunately, tumor cells and T cells in the microenvironment of GBM produce low levels of IFNy, thereby decreasing tumor immunogenicity and disrupting regulatory mechanisms to curtail proliferation and angiogenesis [12]. Like PD-L1, IDO-1/HLA-DR levels could also provide insight into potential response to checkpoint inhibitor immunotherapy in GBM.
More generally, this technology provided us a comprehensive snapshot of the microenvironment of GBM. Myeloid-derived suppressor cells (MDSCs) represent a heterogeneous population of myeloid cells that are present in cancer, inflammation, and infection [13]. The inhibitory effects of MDSCs on innate and adaptive immunity lead to blocking immune surveillance and preventing the immune system from eliminating newly transformed cells. MDSCs can also differentiate into tumor-associated macrophages (TAMs) within the tumor environment. TAMs promote tumor growth, regulate metastasis, mediate response to therapy, and often lead to worse survival outcome. The expression of specific cell surface markers such as CD11b (a marker of macrophages and microglia which regulates leukocyte adhesion and migration to mediate inflammatory response), HLA-DR, and MDSC-specific enzymes such as IDO-1 makes it possible to identify MDSCs [14].
T cells also play a critical role in the immune response and are affected by MDSCs. They can be separated into three major groups based on function: cytotoxic T cells (CD8+), helper T cells (CD4+), and regulatory T cells (T regs). Differential expression of markers on the cell surface, as well as their distinct cytokine secretion profiles, provide valuable clues to the diverse nature and function of T cells. Immunohistochemistry can help to classify a subset of T cells and their activation status by the presence of Ki67, a marker for proliferation, and FoxP3, a marker for immunosuppression through T cell regulation. Many primary tumors have massive infiltration of macrophages, which can make up 30-50% of tumor mass including central nervous system (CNS) tumors like GBM and brain metastases [15]. In general, counts of low CD8+ cells, high CD4+ cells, high T regulatory cells, low IFNy, and high TAMs correlate with a more immunosuppressed microenvironment. The brain uses two types of cells in the immune response: microglia as the first line of defense and macrophages recruited from the tumor periphery or outside the brain. Chen et al. demonstrate that the macrophage marker CD45Hi accounts for 1.5 (+/-3)% of total brain myeloid cell population in naïve mice and 87.5 (+/- 6.2)% in mice with brain tumors [16]; however, CD45Hi levels can only differentiate macrophages and microglia under steady-state conditions and not under inflammatory conditions. This suggests the integral role of macrophage recruitment in the localized tumor immunosuppression in gliomas and provide a framework for investigating other biomarkers that may respond to targeted immunotherapy intervention.
{kind=link}
{kind=link}