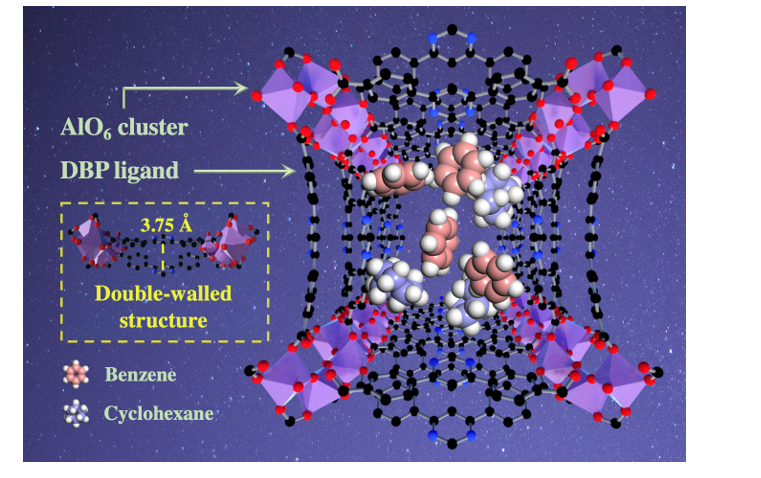
Crystal of ZJU-520(Al) is rod-shaped (Fig. S1), with high purity due to the agreement of the theorical powder X-ray diffraction (PXRD) patterns with the experimental PXRD patterns (Fig. S2). ZJU-520(Al) is in the tetragonal crystal system with I41md space group and lattice parameters (a = b = 36.65 Å, c = 10.56 Å), identified by single crystal X-ray diffraction (SC-XRD) analysis (Table S1-3). Each Al3+ center, in the helical chain of AlO6 cluster, is octahedrally coordinated with O atoms from four DBP2− ligands and two bridging hydroxyl anions (Fig. 1), forming the double-walled structure of ZJU-520(Al) (Fig. 1). The distance of double-wall on ZJU-520(Al) is 3.75 Å (Fig. 1), slightly higher than that of BUT-53(Co), i.e., 3.00 Å, similar to that of BUT-54(Co) to BUT-58(Zn) (Fig. S3) 1. The AlO6 clusters further coordinate to DBP2− ligands, forming the 3D framework (Fig. 1), identified by the slight blue shift of the peaks of carbonyl group at 1589 cm− 1 in Fourier-transform infrared spectra of ZJU-520(Al) from that of H2DBP at 1682 cm− 1 (Fig. S4). ZJU-520(Al) is with two types of 1D channels, named as A with narrow channel and B with wide channel here (Fig. 1), due to the rotation of AlO6 clusters with angle about 11.01° (Fig. S5), compared with the AlO6 clusters of CAU-10(Al) 8, 9. The elemental contents of ZJU-520(Al), including Al, C, O and N, are uniform dispersion and in line with the molecule formula (C21H19AlN3O7) obtained from SC-XRD analysis (Table S1), identified by energy dispersive spectroscope (EDS) mapping and EDS linear scans (Fig. S6).
ZJU-520(Al) has special surface area (SBET) of 2235 m2/g, obtained from nitrogen (N2) adsorption-desorption isotherm at 77 K (Fig. 2a) by Brunauer-Emmett-Teller (BET) method (Fig. S7), in agreement with the theorical specific surface area of 2348 m2/g from Grand Canonical Monte Carlo (GCMC) simulation 10, 11, 12, larger than the reported double-walled MOFs 1 in the range of 849–1128 m2/g (Table S4). For example, the SBET of double-walled ZJU-520(Al) is 1.98 times of that of double-walled BUT-54(Co) (1128 m2/g) and 2.63 times of that of double-walled BUT-58(Zn) (849 m2/g) 1. The total pore volume of ZJU-520(Al) is 0.84 cm3/g (Fig. S8), in agreement with the calculated theorical pore volume of 0.95 cm3/g by GCMC simulation 13. ZJU-520(Al) is a microporous material, identified by its N2 adsorption-desorption isotherm belonging to type I 9, 14, 15 (Fig. 2a) and its pore size distribution (PSD) in the range of 9.26–12.99 Å with pore dimensions centered at 10.96 Å (Fig. S8). It is with two types of microporous channels (Fig. S9) by GCMC simulation, using N2 as probe 16. One channel, named as A, is with the distance of 10.76–16.07 Å (Fig. S10a), while another channel, named as B, is slightly larger with the distance of 11.09–16.67 Å (Fig. S10b), in line with the result of PSD (Fig. S8). The interplanar spacing is 12.83 Å along the (2, 2, 0) direction, observed from high-resolution transmission electron microscopy (HRTEM) image (Fig. 2b), in agreement with the theorical interplanar spacing of 12.96 Å (Table S5). Furthermore, the channels along the (2, 2, 0) direction in the two-dimensional image, calculated by the inverse Fourier transformation from amplitudes extracted from HRTEM image 17, 18, is in good agreement with the framework of ZJU-520(Al) (Fig. 2b).
ZJU-520(Al) is with excellent thermal stability, identified by the thermal gravimetric analysis (TGA) 12, 19 of activated ZJU-520(Al) up to 550 ℃ (Fig. 2c). The quality of ZJU-520(Al) has decreased by 7.78% as the temperature rises to 100 ℃, which should be attributed to the evaporation of water molecules 20, 21. The framework of ZJU-520(Al) also exhibits excellent chemical stability, identified by the N2 adsorption-desorption isotherms (Fig. 2a) and PXRD patterns (Fig. 2d) of ZJU-520(Al) without obvious changes, after being treated with water (pH = 7), HCl solution (pH = 4) and NaOH solution (pH = 12). The excellent thermal-chemical stability of ZJU-520(Al) can be attributed to electronic-withdrawing effect of pyrimidine N atoms on H2DBP ligands (Fig. S11 and Table S6) enhancing the Al – O coordinate bond 22, 23.
ZJU-520(Al) can adsorb trace benzene up to 5.98 mmol/g (Q0.01) at 298 K and P/P0 = 0.01 (Fig. 3a,b), higher than double-walled BUT-54(Co) (Q0.01 = 4.31 mmol/g) 1 and other previously reported benzene adsorbents (Fig. 4a and Table S4), including PAF-1 (Q0.01 = 3.65 mmol/g) 24, ZJU-620(Al) (Q0.01 = 3.80 mmol/g) 4 and UiO-66(CuII) (Q0.01 = 3.92 mmol/g) 25. Isotherms of benzene adsorption on ZJU-520(Al) are quickly increased at trace concentration and reached a plateau at P/P0 = 0.10 (Fig. 4b), indicating the high affinity between adsorbates and framework 26. Even at 308 K, ZJU-520(Al) can keep excellent trace benzene adsorption of 3.63 mmol/g (Q0.01) and rapidly up to 8.09 mmol/g at P/P0 = 0.02 (Fig. 4b). It also has excellent saturation adsorption capacity for benzene (12.07 mmol/g), toluene (6.91 mmol/g), ethylbenzene (3.76 mmol/g), ortho-xylene (4.01 mmol/g), meta-xylene (3.67 mmol/g), para-xylene (4.45 mmol/g) and cyclohexane (5.27 mmol/g) at 298 K (Fig. 3a,b). Besides, ZJU-520(Al) is with excellent regenerate ability, due to benzene adsorption without obvious loss at least 4 times (Fig. 4c) and its framework with integrality due to the PXRD patterns of ZJU-520(Al) without obvious changes (Fig. S12) after cyclical benzene adsorption-desorption experiments.
The isosteric heat (∆H) values of benzene adsorption on ZJU-520(Al) are modestly high and increased in two steps (Fig. 4d), calculated from the isotherms at four temperatures (Fig. 4b). One step is increased at low benzene adsorption (0.00013–1.10 mmol/g) with the ∆H values of 39.68–41.59 kJ/mol owing to the dominance of guest – host interactions 25. Another step is increased at high benzene adsorption (3.56–9.79 mmol/g) with the ∆H values of 40.02–45.54 kJ/mol due to strong guest – guest interactions, such as the packing of benzene molecules 25. The high ∆H values indicates that strong interaction with benzene molecules existed even at low concentration, in agreement with its type I benzene adsorption isotherm (Fig. 3a). Moreover, the ∆H value at benzene loading of 1.00 mmol/g is 41.58 kJ/mol, close to the other MOFs with excellent trace benzene adsorption, such as UiO-66-defect (47.00 kJ/mol) 25, MFM-300 (Sc) (42.00 kJ/mol) 25 and BUT-67(Zr) (55.00 kJ/mol) 24, indicating that it needs moderate energy for regeneration 27.
Grand Canonical Monte Carlo (GCMC) simulations are performed to gain insight into the benzene adsorption sites on ZJU-520(Al). Before GCMC simulation, the partial charges of ZJU-520(Al) are extracted from density-derived electrostatic and chemical (DDEC) method 28, 29 (Fig. S13 and Table S7) for electrostatic interactions. The simulated benzene adsorption isotherm on ZJU-520(Al) is well consistent with the experimental results at low to middle pressures (Fig. S14), but slightly smaller than the experimental results at high pressures, due to the condensation of benzene vapor 30. For example, the theorical benzene adsorption is 6.29 mmol/g at P/P0 = 0.01 and 298 K, in agreement with the experimental value (5.98 mmol/g) (Fig. 3a). There exist two types of benzene binding conformations, i.e., site I near the AlO6 clusters and site II near the N atom of ligands (Fig. 5a,b and Movie S1 for P/P0 = 0.01), due to host – guest interactions (Fig. 4d, in blue area), identified by the slices of calculated potential field. As is also observed for toluene adsorption (Fig. S15, Movies S2). With the P/P0 up to 0.10, benzene molecules also can be adsorbed in the center of channel of ZJU-520(Al) (Fig. 5c), due to strong guest – guest interactions (Fig. 4d, in pink area). Benzene molecules, in channel A and B of ZJU-520(Al), preferentially adsorb in site I (Fig. 5a) due to Al – π interactions 31 and C – H ⋯ X interactions, and then, with the increasing of P/P0, such as up to 0.01, to occupy the site II (Fig. 5b) due to C – H ⋯ N interactions 1, 32. Taking benzene molecules in channel A at P/P0 = 0.01 and 298 K for example, benzene molecules in site I bound to Al atoms of AlO6 clusters through Al – π interactions 31 with the distance of 5.98–8.16 Å (Fig. 5d and Fig. S16a, in bule lines), and also bound to the C – H sectors of ligands through C – H(L) ⋯ π(Bz) interactions 33 with the distance of 3.13–4.95 Å (Fig. 5d and Fig. S16b, in pink lines), where H(L) represents the H atom of DBP ligands and Bz represents benzene molecules. Moreover, benzene molecules also could bind to ligands through C – H(Bz) ⋯ π(L) interactions with the distance of 3.90–5.79 Å (Fig. 5d and Fig. S16c, in green lines), where H(Bz) represents the H atom of benzene molecules. For site II, benzene molecules bound to N atoms of ligands through C – H(Bz) ⋯ N(L) interactions 1 with the distance of 2.63–4.57 Å (Fig. 5d and Fig. S16d, in red lines), where N(L) represents the N atom of DBP ligands. In addition, benzene molecules in site I and site II are all interacted with other guest molecules, through C – H(Bz) ⋯ π(Bz) interactions with the distance of 3.92–5.52 Å (Fig. 5e and Fig. S16e, in purple lines). Therefore, the host – guest interactions and guest – guest interactions drive the benzene adsorption by ZJU-520(Al), even at P/P0 up to 0.01.
The preferential adsorption of benzene molecules in site I (Fig. 5a) also can be explained by the calculated binding energy (–69.68 kJ/mol) between benzene molecules and ZJU-520(Al), which is higher than that in site II (–46.23 kJ/mol). The binding energy is calculated based on the low-energy structure role of ZJU-520(Al) with adsorbed benzene molecules, using the density functional theory (DFT) calculation method 34, 35, 36, 37. Benzene molecules in site I have stronger charge density than that in site II, making benzene molecules preferentially adsorb in site I rather than site II especially at trace concentration (Fig. 5a,b), due to the electron accumulation 38 in the center of benzene molecule (site I) and depletion 1 that of benzene molecule (site II and center) (Fig. S17).
Separating benzene (Bz) from cyclohexane (Cy) is a very difficult work in industry such as nylon production 21, 39, due to similar boiling points of Bz (353.25 K) with Cy (353.85 K) 40 and similar kinetic diameters of Bz (5.90 Å) with Cy (6.20 Å) 41 (Fig. 6a). Adsorption of benzene by ZJU-520(Al) (5.98 mmol/g), at 298 K and P/P0 = 0.01, is 2.56 times that of cyclohexane (2.33 mmol/g) (Fig. 3a). The ideal adsorbed solution theory (IAST) selectivity 27, 42 of Bz/Cy mixture at vapor volume of 50/50 by ZJU-520(Al) is 29.86 (Fig. S18), exceeding that by CUB-5 (4.2) 43, MFOF-1 (5.3) 44 and Mn-TCNQ-bpy (15.2) 44. In the breakthrough experiments of Bz/Cy (5/95, in red line) and Bz/Cy (1/99, in black line) mixture vapor (Fig. 6b), pure cyclohexane elutes with high-purity firstly, whereas trace benzene still is adsorbed in the fixed bed and retained with a longer time course (Fig. 6b), further indicating its excellent trace benzene separation from mixed vapors of Bz/Cy. ZJU-520(Al) is also with excellent recyclability for Bz/Cy separation at least 4 times (Fig. 6c). Excellent Bz/Cy separation of ZJU-520(Al) could be attributed to the higher adsorption affinity with benzene than cyclohexane molecules, identified by the higher ∆H of benzene than that of cyclohexane at trace adsorption of benzene and cyclohexane less than 3.89 mmol/g (Fig. S19a), calculated from benzene isotherms (Fig. 4b) and cyclohexane isotherms (Fig. S19b) at various temperatures. Using multicomponent GCMC simulations of benzene and cyclohexane adsorptions 45, the adsorption site of cyclohexane is near the AlO6 cluster due to C – H(Cy) ⋯ N(L) interactions with the distance of 3.20–5.54 Å (Fig. 6d and Fig. S20a, in black lines) and C – H(Cy) ⋯ π(L) interactions with the distance of 2.59–4.83 Å (Fig. 6d and Fig. S20b, in orange lines), where H(Cy) represents the H atom of cyclohexane molecules, while the adsorbed benzene molecules interact with ZJU-520(Al) through multiple interactions, including Al – π interaction with the distance of 5.67–6.73 Å (Fig. 6e and Fig. S21a, in blue lines), C – H(L) ⋯ π(Bz) interactions with the distance of 3.11–6.00 Å (Fig. 6e and Fig. S21b, in pink lines), C – H(Bz) ⋯ π(L) interactions with the distance of 3.21–4.91 Å (Fig. 6e and Fig. S21c, in green lines), C – H(Bz) ⋯ N(L) with the distance of 3.09–6.37 Å (Fig. 6e and Fig. S21d, in red lines) and π(Bz) ⋯ π(L) interactions with the distance of 3.81–7.33 Å (Fig. 6e and Fig. S21e, in brown lines). Therefore, ZJU-520(Al) exhibits more multiple interactions with benzene than that with cyclohexane, resulting in the excellent Bz/Cy separation of ZJU-520(Al), even at trace benzene concentration.
{kind=link}