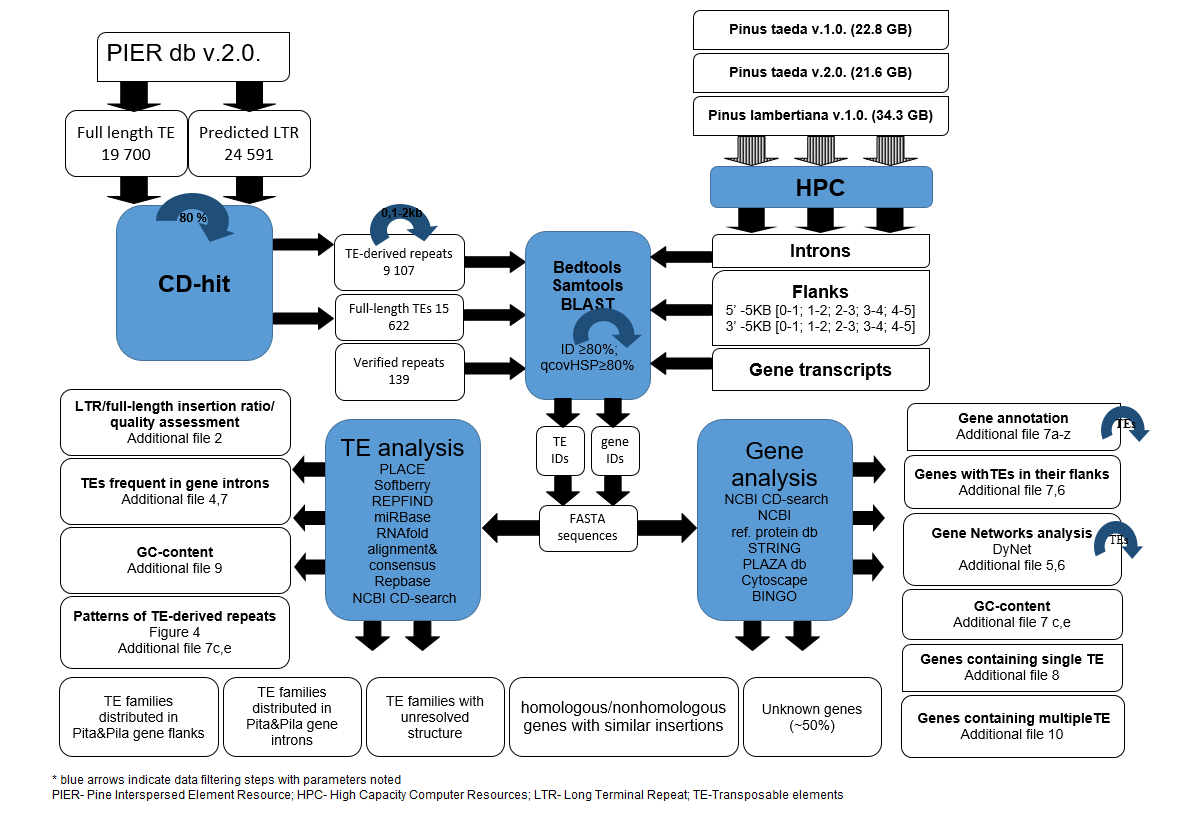
Automated TE detection relies on several strategies, such as searches for sequences with homology to known elements, de novo evaluation of repeated elements, analysis of the presence of specific structural features, and combined techniques [93]. In the genome assembly process, repeated and highly similar sequences like TEs are the source of errors and gaps [94, 95]. Properties of plant genomes, such as multiple gene families, pseudogenes, and chromosomal and plastid genome duplications, further complicate the process of genome assembly and annotation [96, 97]. Typically, gymnosperms are characterized by large genomes with proliferated TEs, high levels of heterozygosity, a constant chromosome number, and very rare polyploidy events [58, 98]. Several conifer genomes have been sequenced using short-read assembly methods [60, 62, 66, 99]; the P. taeda v.2.0 genome currently has the highest quality conifer genome regarding scaffold length. Version 2.0 was improved by merging small contigs; while the first version of the P. taeda genome contained 16.5 million contigs, the second version contains only 2.9 million [63]. However, the average PacBio read length used to reconstruct genome v.2.0 was 9,665 bp with 12x coverage, which is shorter than many RLXs. The mean TE length in the PIER database was 6,273 bp (median 5,383 bp) but 3,046 TEs were longer than 10 Kbp. With the availability of better quality or longer-read genome assemblies, TE identification should be repeated de novo. Genome annotation of conifers is an ongoing process. Indeed, our study revealed errors in annotation files and datasets, where gene IDs of transcripts did not coincide with genomic sequences bearing identical gene IDs, and genes had noticeably varying intron lengths in different genome versions. Some additional errors involve automated annotation, nested repeat annotation, defined pools of identical reverse transcriptase domains as different genes, and intense masking of ambiguous regions, which are not discussed sufficiently in associated publications. Consequently, comparison of genomes assembled with different approaches and of varying quality should be performed with caution to avoid errors in interpreting the results. Additionally, widespread gene capture by TEs has often been described for large plant genomes [42, 100–102] and this phenomenon could introduce additional errors in short-read genome assemblies [102]. Considering the high copy number and the structural and functional differences from protein-coding genes, it is advisable to annotate TE-associated sequences in separate data sets even though many TE families bear ORFs. Despite the fact that many conifer TEs only show weak similarity to annotated TE domains, complexity was effectively reduced in the P. lambertiana high-quality gene set [64]. Evaluation of the only prevalent MITE element in the flanking regions of initial genome versions demonstrates the importance of read length in assembly and the association of TEs to particular genome locations.
Diverse conifer RLXs sharing high partial sequence similarities may not be classified in one family by full-length alignments used in bioinformatic studies, resulting in inflated family counts [66]. Due to the high diversity of conifer TE sequences and patchy distribution within short read assemblies, we found that identification of short repeated regions could overcome these problems and more efficiently identify prevalent TE families. The use of clustered TE-derived repeats extracted from automatically predicted nested regions allowed not only for the identification of RLX, but also internal regions of other TEs. The use of TE-derived repeats allowed simple statistical tests of distribution relative to distance from genes. RLXs are the most widely distributed TE class in plants and gymnosperm genomes [32, 60, 66, 103]. LTRs contain important regulatory signals that can influence gene expression even if the body of the element is deleted [22, 25, 104]. The PIER v.2. database entries contain nested TEs from automated predictions, including repeated internal regions of different TEs. Chimeric TEs that could evolve from two RLXs by template switching [43] and display signals of independent transposition have been identified in other plant genomes, e.g. Veju [105] and BARE-2 [106]. TE structures with several LTRs are found in plant genomes, for example, 13 Cassandra elements in pear contain 3 LTRs [107]. Considering the large conifer genome sizes, such chimeric structures may also be found in conifer genomes. Therefore, each high-frequency repeat should be verified and the true full-length structure and TSDs should be identified. Unfortunately, the pine genomes contain many low-copy-number TEs, and if all copies are masked, full-length structures or TSDs cannot be resolved. Additionally, similarity to known proteins and GO annotations are available for only approximately half of all pine genes, but species-specific genes could be more important in adaptation of species. Therefore, the data evaluated and the results obtained from this study could be expanded with improvement of genome quality, annotation, or other associated information.
In the current study, we identified several TE families that unite many pine stress-responsive genes and contain potentially important gene regulatory signals. The DTX184 DNA TE could provide microRNA target sites or produce microRNA, or both. However, further analysis is required to determine if these sequences represent microRNA precursors or, alternatively target sites [108]. This DNA TE insertion was found in the second intron of the P. taeda NPR1 gene, a key regulator of systemic acquired resistance [109], the PSMD4 gene coding for the 26S proteasome (involved in the ATP-dependent degradation of ubiquitinated proteins), ubiquitin thioesterase OTU1 (plays an important regulatory role at the level of protein turnover by preventing degradation), S-formylglutathione hydrolase (detoxifies formaldehyde), a PHD finger protein ALFIN-LIKE 4 (a histone-binding component that recognizes H3K4me3), and other important genes. Based on the presence of short consensus sequences of the DTX184, approximately 200 genes could be involved in this network in the P. taeda genome. In P. lambertiana, the similarity of DTX184 with 143 high-quality genes with high coverage was found, many of which are important genes coding for transcription factors, chromatin modification enzymes, protein kinases, and receptors. The short MITE3321 family identified in proximal gene flanking regions and introns could provide TATA boxes and several ARR1, DOF, W-box, GT and MYB-binding sites, which are important signals in plant transcription activation and stress-response regulation [110–112]. Ten different ARR1 binding sites (7 on positive and 3 on negative strand) were present in the consensus sequence of P.taeda MITE3321 element. The transcription factor-type response regulator ARR1 directs transcriptional activation of the ARR6 gene, which responds to cytokinins without de novo protein synthesis [113]. Cytokinins are an important class of phytohormones that are involved developmental processes and growth [114–116], as well as in defense responses [117]. The (AG)4A motif is one of the most common TFBS for plant promoters [118], that regulate light-responsive phototransduction processes in plants [119]. The Copia-1813 identified in this study contain longer AG-rich tracts, this RLX family is distributed in pine gene introns and several flanks and might form responsive gene network. Genome-wide approaches in mammalian genomes demonstrate that TEs contribute to rewiring and selection of gene networks [120, 121]. Approximately one-sixth of rice genes are associated with TEs [122]. Comparative analysis of three inbred maize lines revealed that the expression of 33% of stress-responsive genes could be attributed to regulation by TEs [123]. Various maize TEs contain approximately 25% of all DNase I hypersensitivity sites within the genome, which are associated with open chromatin and cis-acting elements and are therefore essential transcriptional regulators [124]. In contrast to mammals, plant genes more commonly contain longer introns, which are expressed at higher levels [125, 126]. However, in recent studies, expression in different tissues or expression breadth has also been considered and plant genes expressed across a wide range of tissues or conditions were found to have a higher intron density [127, 128]. In P. taeda and P. lambertiana, extensive transcriptome data is not currently available, therefore the role of TE insertions within introns in regulation of gene expression requires additional investigations. In the Copia-1813 RLX gene network identified in this study, formation of specific patterns or intronic genotypes was supposed. TE patterns could influence gene availability, responsiveness, stability, or higher order organization structures in the nucleus. Two genes with identical TE insertion patterns are involved in pre-mRNA maturation and splicing, while other genes with identical TE insertion genotypes are linked to protein metabolic processes and Golgi body homeostasis. Further investigation will enable more thorough analyses of these processes. The function of genes where multiple TEs were identified within introns (e.g. potassium channel coding genes and other receptors, protein kinases, cytochrome genes) suggests involvement in the maintenance of cell homeostasis under stress conditions. These genes were found to have many co-occurring GO terms, indicating that gene products are involved in many cellular processes and these genes may be expressed in a broad range of conditions. We suggest that genes with many different types of TEs could act as node genes that are functional or stable across a range of conditions and could be important in early defense responses and rapid metabolome switching. This is also supported by the discovery of several homologous genes with large introns in both pine species. Cases of independent transpositions of different RLX families into homologous genes in varieties of differing origin resulting in similar phenotypes have been reported [22]. Additionally, the TE insertion patterns in investigated pine introns were found to have lower average GC content (39%) than nearby transcripts. The GC content of gene transcripts in the studied gene networks in P. taeda and P. lambertiana were comparable (44%) and higher than the reported genome average of 38% [90, 91]. A lower GC content in plant gene introns has been reported for other plant species [129, 130], indicating that intron sequences may have a more relaxed DNA conformation and are more accessible to transcription and other regulatory factors [131, 132].
It remains unclear if TE sequences have insertional preferences, but it has been reported that a relaxed chromatin configuration promotes TE insertions and leads to an increased mutation probability in tissue- and stage-specific genes [133, 134]. In this aspect, TEs should be randomly distributed in different stress-responsive gene non-coding regions (flanks or introns). The non-autonomous DNA TE MITE3321 element insertions were statistically significantly overrepresented in the proximity of pine genes (0–2 kb), a distance over which linkage equilibrium extends in P. taeda [135]. MITEs are preferentially located within gene regions of many plants and could influence gene expression [46, 47, 136–138]. There is evidence that some MITE insertions located close to gene promoters could also downregulate gene expression [139]. MITE3321 was frequently inserted also in the introns of genes of the studied pine species. However, in this study, no genes with several MITE3321 insertions in its different non-coding regions (flanks and introns) were identified. This distribution suggests formation of differentially regulated gene sub-networks, depending on the location of MITE insertions. For example, it was determined that many genes containing MITE3321 in flanking regions were involved in the regulation of developmental processes and cell division, but genes having MITE3321 in their introns were associated with immune responses and cell-wall biosynthesis, among other activities. If MITE3321 cis-acting elements in the more accessible introns help activate a specific network of genes, then non-coding RNA products of splicing from stress-responsive genes could be involved in feedback loop mechanisms for blocking transcription of genes with proximal MITE3321. In this example, identical TEs could have a downregulatory effect on developmental gene transcription, switching to an energy-saving mode, while at the same time increasing transcription of defense genes. Similar strategies could be highly advantageous for the rapid activation of defense responses and switching of metabolic functions. Additionally, in the current study, MITE3321 insertions were found in both analyzed pine species, belonging to separate subgenera, suggesting similar distributions also in other pine species. Therefore, MITE3321 could be a useful molecular marker for genotyping of pine species, as shown for MITEs in other plant species [48, 140, 141].
TE distribution is linked with speciation [142, 143]. In pines, transposition activity accounts for the period following the divergence of the pine subgenera Strobus (P. lambertiana) and Pinus (P. taeda) and speciation [62, 65, 67, 68, 144]. Only a few homologous genes in both pine species were found with similar TE insertions, indicating that differences in TE family distribution between species are also found in more conservative gene regions. Insertion of DTX184 in homologous genes of both species was found in PSMD4, a 26S proteasome non-ATPase regulatory subunit gene (40% query coverage, 98% sequence identity of transcripts). Insertion of the ancient Gypsy RLX IFG [67] was found in three homologous P. taeda and P. lambertiana gene pairs, surprisingly, all encoding different protein kinases. However, some of the IFG insertions were located in different introns of kinase genes and therefore could also represent independent transpositions. Analyzed IFG sequences contained masked regions and therefore the age of the insertions was not evaluated. Notably, insertions of other TEs were also frequently found in different receptor-like protein kinases and tyrosine protein kinases in our study. Protein kinase genes belong to one of the most proliferated gene superfamilies in plants, whose members are linked with a range of key metabolic and various plant-specific adaptation processes [145–148].
In conclusion, the source of TE sequences expressed in response to stress conditions could be the transcription of introns of many stress-responsive genes, which could explain the observed highly correlated expression levels of RLX families within individuals [70]. Transfer of information about TE insertions in gene regions to non-model pine species is complicated, as common TE families were revealed, but they are generally located in non-homologous genes. This highlights the need for additional studies and sequencing of species of interest to investigate TE-associated polymorphisms, such as in P. sylvestris, which is an important species in northern Europe. In the two analyzed pine species, TE insertions were more often found in gene introns but less commonly in gene flanking regions, similar to other plant species [149–152]. Insertions of TEs in gene regions are associated with various epigenetic mechanisms, but it remains unknown how some actively transcribed plant genes are coping with large introns [139]. Cultivation and plant breeding reduces genetic diversity and fitness to environmental stresses [153]. A recent whole-genome comparison of wild and cultivated rice species revealed depletion of intronic TE insertions in cultivated species [154]. Gymnosperms are outcrossing species that produce large quantities of pollen and seeds, generating a genetically diverse germplasm pool for subsequent natural selection of highly adaptable seedlings. Pine species are known for their strong adaptation to local growing conditions [155–158]. This study demonstrated the increased accumulation of TE sequences in stress-responsive gene introns and the probability of rewiring them in responsive networks interconnected with node genes containing multiple TEs. Preferential TE insertion in open chromatin has been reported for tissue-specific genes [133], hence similar mechanisms followed by sustained natural selection could drive the accumulation of fitting TEs in gene non-coding sequences of plants. The inclusion or exclusion of genes from TE-mediated networks is an efficient way means of dynamic change in response to various environmental factors, including changing host-pathogen interactions and unresolved layered processes in plants such as communication and signaling organization. Therefore, many such regulatory influences could lead to the adaptive environmental response clines that are characteristic of naturally spread pine populations [159–162].
{kind=link}