Bioinformatic detection of branch points among the physiological and alternative splice acceptor sites
In this study, two sets of 3’ss data were used, 3’ss described in Ensembl dataset and alternative 3’ss with their expression data from RNA-seq analyses (Table 2). The running times showed that BPP is one of the faster tools and Branchpointer one of the slower tools (Supplementary Figure S3).
We first retrieved 264,787 Ensembl 3’ss from the Ensembl data. Adding to these 3’ss, 114,603,295 random AGs were used as control data (see the “Methods” section for details). Thus, we collected 114,868,082 3’ss. ROC curve analysis was then performed for SVM-BPfinder, BPP, LaBranchoR and RNABPS on the set of Ensembl 3’ss, as illustrated in Figure 2A. Table 3 shows the levels of accuracy, sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) derived from these ROC curve analyses. In terms of the area under the curves (AUC), the score provided by BPP exhibited the best performance (AUC = 0.818). However, Branchpointer presented the highest performances with an accuracy of 99.49 % and PPV of 30.06 %. Thus, Branchpointer was the most stringent of the bioinformatic tools for detecting putative BPs upstream of Ensembl 3’ss. Indeed, SVM-BPfinder, BPP, LaBranchoR and RNABPS detected putative BPs for each Ensembl 3’ss and random AGs. For these 4 tools, the best accuracy to distinguish Ensembl 3’ss from random AGs was reached by BPP (75.23 %). Overall, 74,539,834 3’ss had a BP predicted by at least one tool. The maximum overlap of predicted BPs was observed between LaBranchoR and RNABPS (28.63 %; 21,337,483/74,539,834 3’ss) (Supplementary Figure S4). The percentage of 3’ss with BP predicted by the five tools was 0.15 % (111,937/74,539,834). Seventy-five percent (83,892/111,937) of these 3’ss were Ensembl 3’ss (Supplementary Figure S5).
Among the alternative junctions of whole transcriptome analysis, 51,986 alternative 3’ss were identified (see the “Methods” section for details and Supplementary Figure S6), to which we added the same number of control 3’ss. In all, we had 2 subsets of 51,986 (103,972) acceptor sites for whole transcriptomic data (Supplementary Table S1). The SpliceLauncher analysis revealed that 99.5 % of splicing junctions (51,703/51,988, data not shown) did not have a significant expression difference across the different cell culture conditions and the different variants. The relative expression of the alternative 3’ss appeared to follow a log-normal distribution (Shapiro-Wilk p-value = 0.09 and Supplementary Figure S7). From these data, Branchpointer outperformed all tested tools for detecting putative BPs (Table 4). Indeed, the AUC of the three tools, SVM-BPfinder, BPP, LaBranchoR and RNABPS, did not perform above 0.612 (RNABPS) (Figure 2B). Branchpointer showed the best accuracy of 65.8 % on the alternative splice sites. Furthermore, this tool demonstrated a similar specificity with the Ensembl and RNA-seq data, 99.6 % and 99.5 %, respectively. However, on the whole transcriptome data, the sensitivity decreased by more than 60 % (from 95.5 % to 32.1 %) (Table 3 and Table 4). The alternative 3’ss and control 3’ss had BPs predicted by at least one of the tools in 91.2% (94,806/103,972). The maximum overlap was observed between the four tools SVM-BPfinder, BPP, LaBranchoR and RNABPS (7,227/94,806 3’ss). More than 95 % of 3’ss with a BP predicted only by Branchpointer were alternative splice sites (Supplementary Figure S8). In a paired comparison, the two tools LaBranchoR and RNABPS displayed a maximum overlap of 34.57 % (32,777/94,806 3’ss) with common BPs (Supplementary Figure S4).
We compared the expression of alternative sites, from RNA-seq data, with and without the presence of a putative BP predicted by the bioinformatic tools (see the “Methods” section for details). This analysis revealed that 3’ss with a predicted BP were significantly more expressed than 3’ss without a predicted BP, regardless of the bioinformatics tool (Figure 3). The greater difference of expression was observed for Branchpointer. The average expression was 34.00 % and 1.35 %, for alternative 3’ss with Branchpointer-predicted BP or not, respectively. In the subgroup of 3’ss with a predicted BP, the Branchpointer score was not correlated with the expression of these sites (R² =0.00001, p-value = 0.24). The other bioinformatics tools presented a weak correlation between their score and the expression (Supplementary Figure S9). Among SVM-BPfinder, BPP, LaBranchoR and RNABPS, the best correlation was obtained with RNABPS (determinant coefficient (R²) = 0.0062, p-value = 4.14x10-70).
Bioinformatic prediction of splicing effect for variants in the branch point area
The last set of data was a collection of experimentally characterized potentially spliceogenic variants mapping within BP areas (see the “Methods” section for details), n = 120 variants among 86 introns in 36 different genes (Table 2 and Supplementary Table S2). Part of this collection was obtained from unpublished data (n = 62 variants). From the 120 variants, 38 (31.7 %) were found to induce splicing alteration, and were therefore considered as spliceogenic, whereas 82 (68.3 %) did not show splicing alterations under our experimental conditions. Figure 4 indicates the repartition of the 120 variants within the corresponding BP areas and their impact on RNA splicing. The 38 spliceogenic variants were identified in 30 different introns; 22 variants induced exon skipping, 10 variants caused full intron retention and six remaining variants activated the use of another cryptic 3’ss located up to 147 nt upstream of the 3’ss and 38 nt downstream of the initial acceptor site (Supplementary Table S2).
After the prediction of BPs for each intron affected by the variants, we analyzed the distribution of each variant according to the position of the predicted BP (Supplementary Figure S10). First, we assayed the different size motifs to classify variants (see the “Methods” section for details). The best common motif was the 4-mer starting 2 nt upstream of the A and 1 nt downstream (Supplementary Figure S11), that corresponds to the motif TRAY. For this size motif, BPP presented the best accuracy with 89.17 % and LaBranchoR had the lower performance with an accuracy of 78.33 % (Table 5). Branchpointer did not predict a BP for the intron 24 of BRCA2 gene causing a missed data point, corresponding to BRCA2 c.9257-18C>A variant.
As shown in Supplementary Figure S10, variants affecting splicing were mostly located at putative branch point positions 0 (the predicted branch point A) and -2 (the T nucleotide 2 nt upstream of the branch point A itself). BPP pinpointed the highest number of spliceogenic variants in these positions. More precisely, splicing anomalies were detected for all of the ten variants occurring at position -2, and for 15 out of 18 variants predicted to be located at the branch point A. The three remaining variants predicted by BPP to alter the branch point A position (BRCA1 c.4186-41A>C, MLH1 c.1668-19A>G and RAD51C c.838-25A>G), and not experimentally validated, were also predicted to alter a BP adenosine by SVM-BPfinder while Branchpointer and LaBranchoR placed these variants outside BP motifs.
Next, we assessed the discriminating capability of each tool, including HSF, by calculating delta scores, to identify splicing defects from BP variants (Figure 2C). In terms of delta score, SVM-BPfinder outperformed the other tools with an AUC of 0.782. From this ROC analysis, we identified an optimal decision threshold (see the “Methods” section for details) of -0.136, i.e. the variants were predicted as spliceogenic if the variant score was less than 13.6 % of the wild-type score. The performances achieved with this threshold are reported in Table 6. SVM-BPfinder reached the maximum accuracy of 81.67 %.
The achievement of cross-validation, from the logistic regression model, highlighted the performance of combination of the BPP and Branchpointer tools (see the “Methods” section for details). This model was to infer variants as spliceogenic if they occurred within a TRAY 4-mer BP motif predicted by both BPP and Branchpointer. Although this combination was mostly found in the 1,000 simulation, this model appeared in only 26 % of these simulations (see Supplementary Figure S12). The likelihood ratio test between this model and a model with only the BPP tool was not systematically significant, with 60.1 % of simulations having p-value above 1 %. This approach also showed that for a variant in intron with different and non-overlapping predicted BP sites by BPP and Branchpointer, the model could not provide prediction of potential spliceogenicity. We continued the cross-validation without the positions of predicted BP for all tools except BPP. However, the delta scores of other tools did not improve the model, as the majority of simulations converging to BPP-alone model (Supplementary Figure S13). Thus, the analysis revealed that the position of the BPs predicted by BPP alone was the optimal model.
{kind=link}
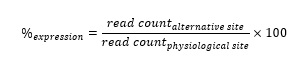{kind=link}