1. Modulation of flotillin-1 palmitoylation alters its stability and membrane localization.
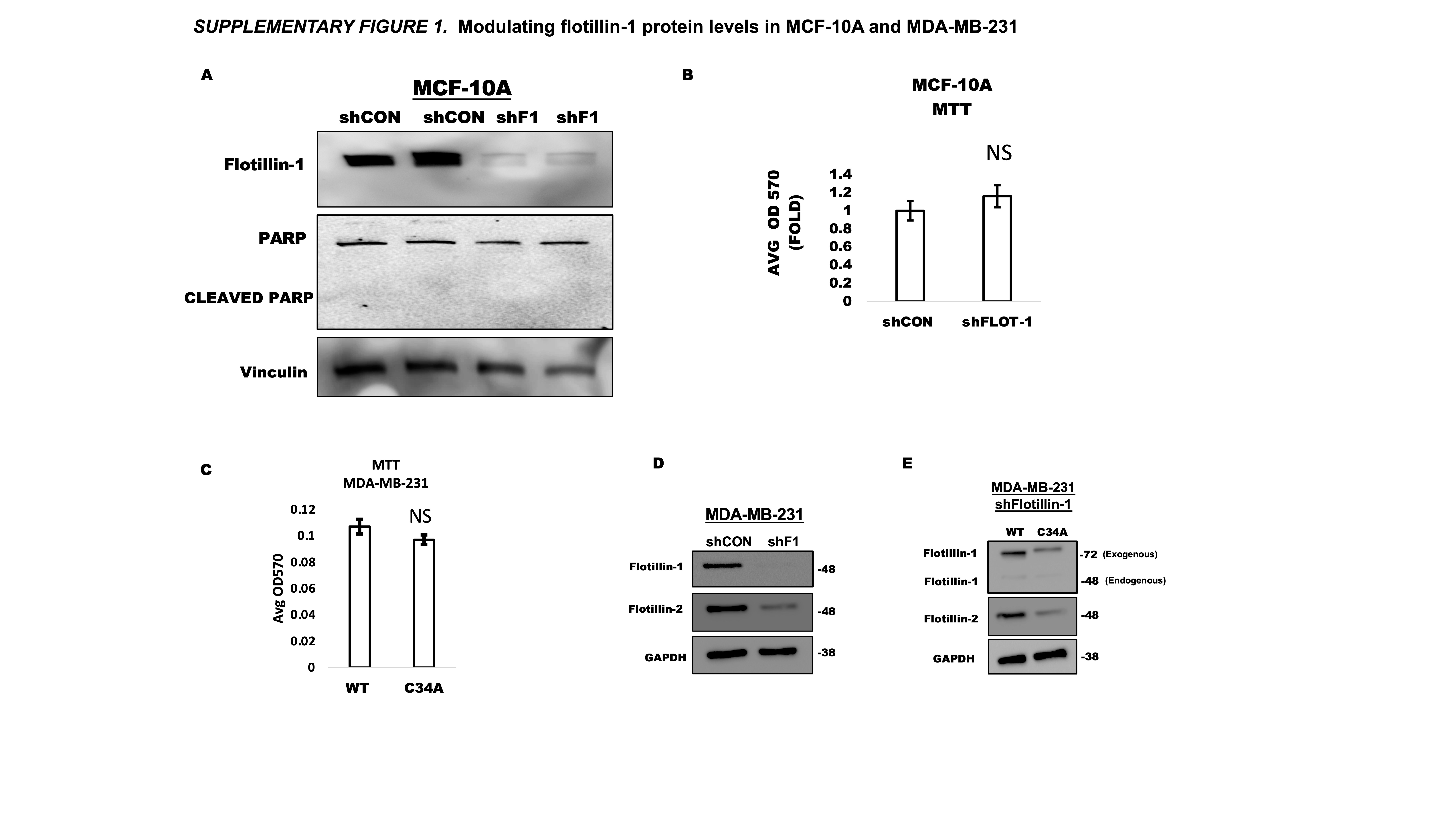
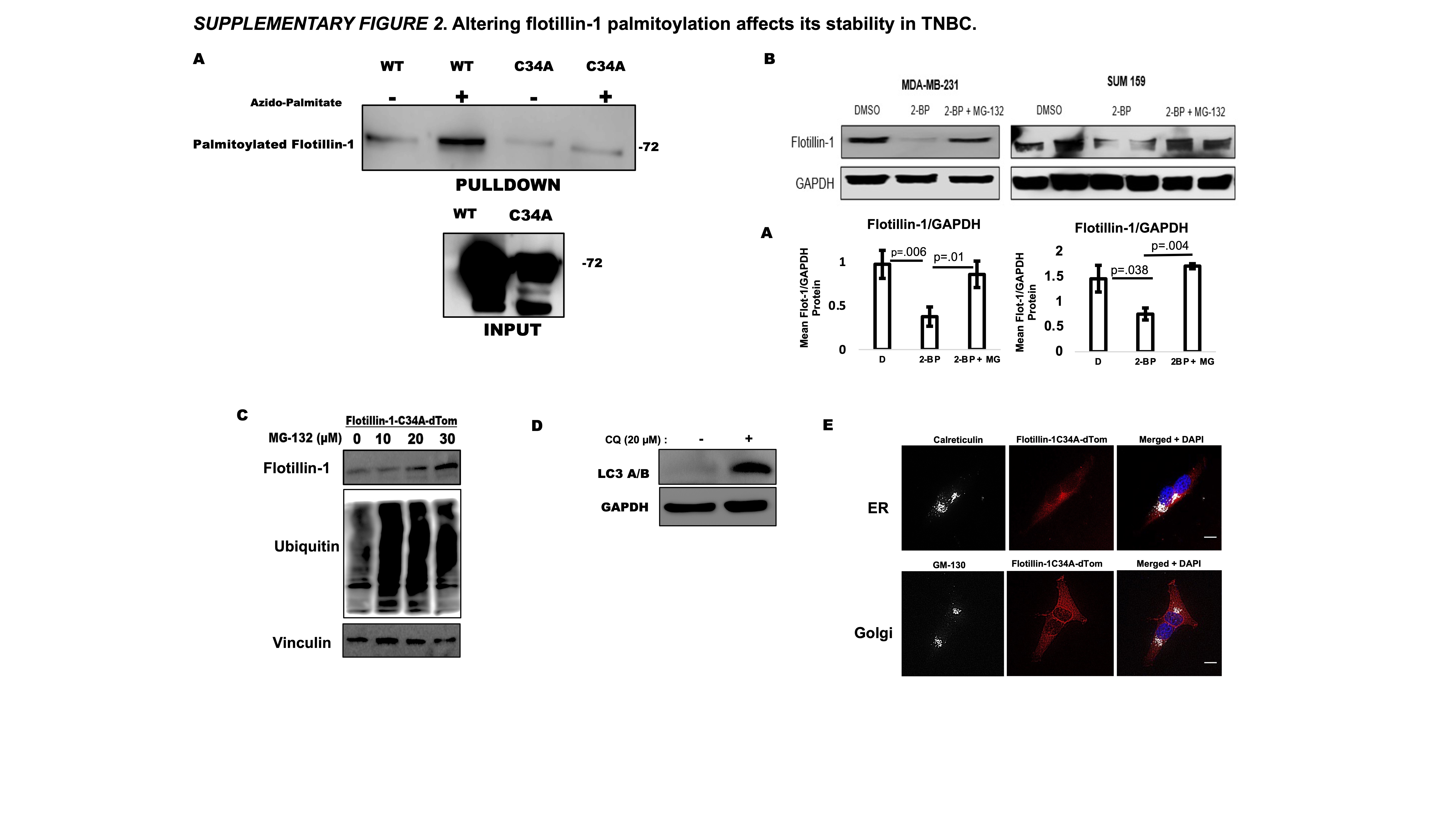
Flotillin-1 is highly expressed in triple negative breast cancer (TNBC) and its expression is associated with metastasis (6, 18). We further confirmed its high expression in TNBC by probing for flotillin-1 protein in a panel of TNBC cell lines, of which revealed high flotillin-1 expression compared to that of luminal (ER+), MCF-7, and non-cancerous (NC) MCF-10A cells (Fig. 1a). Using MCF-10A cells to represent non-cancerous cell populations, we depleted flotillin-1 protein even further through RNAi and did not observe any apoptosis or altered cell viability compared to control, as measured by cleaved-PARP and MTT assay, respectively (Supplementary Fig. 1 a,b). With the preponderance of studies demonstrating the role of flotillin-1 protein expression in cancer progression and metastasis (4, 8, 9), we sought to develop a model to study the effects of its palmitoylation in the context of TNBC metastasis. To this end, we constructed flotillin-1 palmitoylation defective mutants by mutating its palmitoylated cysteine 34 reside to an alanine, a mutation previously used to study its cellular localization (2). Both the floillin-1 wild type (WT) and palmitoylation defective (C34A) constructs were cloned into an eGFP vector to generate GFP fusion proteins (Fig. 1b). We further confirmed the palmitoylation defect through click chemistry and GFP western blotting in HEK 293T cells (Fig.1c). MDA-MB-231 TNBC cells were chosen as the host cell line for the constructs given their high expression of flotillin-1 protein and their ubiquitous use in xenograft models of TNBC metastasis. To avoid any complications with the high level of endogenous flotillin-1 expression, we transduced these MDA-MB-231 cells with a 3’ untranslated region (UTR) targeted shRNA against endogenous flotillin-1 (shF1), which allows for the re-expression of flotillin-1 cDNA (Supplementary Fig.1, d). Upon re-expression of C34A flotillin-1 in shF1 cells, we observed a surprising reduction in ectopically expressed flotillin-1 C34A protein compared to WT (Fig. 1d). To ensure these results were not due to transcription discrepancies between the two constructs, we performed RT-PCR for flotillin-1 mRNA between flotillin-1 WT and C34A expressing cells and found no difference in mRNA levels, suggesting the loss of protein in the C34A flotillin-1 was post-transcriptional (Fig. 1e). For further analysis, we generated stably expressing WT and C34A flotillin-1 cells by transducing shF1 MDA-MB-231 cells with flotillin-1 WT or C34A lentiviral constructs, which still harbored the C34A palmitoylation defect after stable integration and did not display any changes in cell viability (Supplementary Fig. 1a,c). Numerous studies have demonstrated flotillin-1 and 2 protein expression to be dependent on one another (18, 19, 20, 21, 22). Commensurate with these findings, we observed reduction in flotillin-2 protein upon both flotillin-1 depletion and re-expression of flotillin-1 C34A protein (Supplementary Fig 1 d,e). Though we can still visualize and detect flotillin-1 directly through specific antibodies, the role of the additional loss of flotillin-2 on any phenotypic effects observed cannot be ruled out. Palmitoylation can protect proteins from degradation by altering their stability (23, 24). We next asked if the observed protein loss in the flotillin-1 palmitoylation defective construct was due to a decrease in its protein stability. To assess the stability of flotillin-1, we performed a cycloheximide (CHX) chase assay, which revealed an accelerated degradation of the flotillin-1 C34A protein compared to WT (Fig 1f). Palmitoylation of proteins can prevent both lysosomal and proteasomal degradation (23, 25), which motivated us to explore the method in which the flotillin-1 C34A protein degradation was occurring. To discern how flotillin-1 C34A protein was being degraded, we performed rescue experiments with empirically validated (Supplementary Fig. 2 c,d) concentrations of proteasomal (MG-132) or lysosomal (Chloroquine, CQ) inhibitors after depleting flotillin-1 C34A protein with CHX. Flotillin-1 C34A protein levels were significantly restored upon MG-132 treatment, while chloroquine failed to restore flotillin-1 C34A protein (Fig. 1g). The flotillin-1 WT expressing cells were also included as a control and did not display any significant protein alterations with the same conditions (Fig. 1g). The method used for detecting protein palmitoylation with click chemistry requires the enrichment of palmitoylated proteins with streptavidin. To ensure the loss of palmitoylation observed with the flotillin-1 C34A mutation was not due to a decrease in available protein for streptavidin enrichment, we repeated the click chemistry labeling and enrichment method with MG-132 treatment, which restored flotillin-1 C34A protein levels without affecting its palmitoylation status, further confirming its palmitoylation defect (Fig. 1h). To ensure these observations were not specific to ectopic expression of flotillin-1, we treated MDA-MB-231 and SUM 159 TNBC cell lines with a global palmitoylation inhibitor (2-bromopalmitate, 2-BP) with or without MG-132. 2-BP treatment significantly decreased endogenous flotillin-1 protein expression in both cell lines, which was effectively restored upon treatment with MG-132 (Supplementary Fig. 2b). Flotillin-1 palmitoylation was initially studied based on its involvement in its plasma membrane trafficking (2). To assess if the palmitoylation defect was also altering flotillin-1 plasma membrane localization, we performed immunofluorescence in MDA-MB-231 cells expressing a wild type flotillin-1 fused to eGFP and flotillin-1-C34A fused to dTomato. Counter staining with the plasma membrane marker, Na+/K+ ATPase, revealed in a high colocalization with the wild type flotillin-1, compared to the palmitoylation defective flotillin-1 (C34A), even when the protein expression was restored with MG-132 treatment (Fig. 1h). The observed altered flotillin-1-C34A plasma membrane localization prompted us to determine if the flotillin-1 palmitoylation defect was modifying its ability to exit out of the endoplasmic reticulum (ER) or Golgi, given the role of palmitoylation and proper protein processing (2, 26, 27). Counterstaining with ER (Calreticulin) and Golgi (GM-130) specific antibodies revealed no ER or Golgi retention of flotillin-1 C34A (Supplementary Fig. 2e). Collectively, the current results demonstrate an attenuated protein stability and plasma membrane localization upon flotillin-1 palmitoylation modulation.
2. zDHHC5 contributes to the stability of endogenous flotillin-1 by preventing its proteasomal degradation.
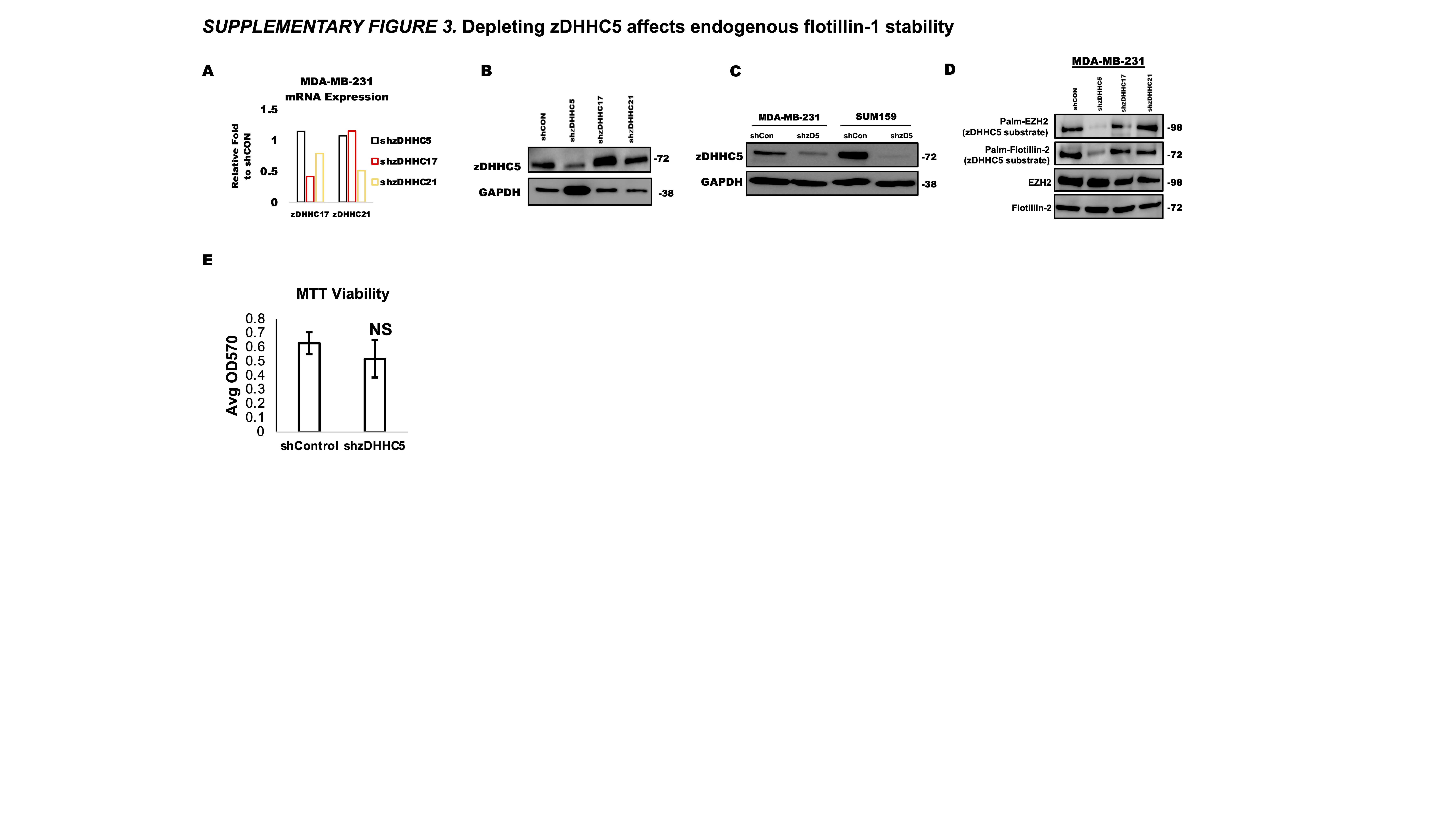
Palmitoyl acyltransferases or zDHHCs are the enzymes responsible for transferring palmitoyl-CoA to target proteins (25, 28, 29). To identify the zDHHC enzyme responsible for palmitoylating endogenous flotillin-1, we performed a mini shRNA screen against the three highest expressing zDHHC enzymes in breast cancer based on data from the human protein atlas (Fig. 2a). After confirming the specificity and knockdown efficiency of the shRNAs by RT-PCR or western blotting, we analyzed flotillin-1 palmitoylation levels in the different stable zDHHC knockdown cell lines (Supplementary Fig. 3 a,b , Fig. 2b). zDHHC 5 knockdown substantially reduced flotillin-1 palmitoylation status compared control, zDHHC 17 and 21 shRNA expressing cells. These findings were also confirmed in MDA-MB-231 and SUM-159 TNBC cell lines without any observed cell viability alterations upon zDHHC5 silencing (Fig. 2d, Supplementary Fig. 3c, e). The palmitoylation sate of two additional zDHHC5 substrates (EZH2 and flotillin-2) were also found to be substantially decreased in the zDHHC5 silenced cells compared to the other conditions (Supplementary Fig. 3d) (28, 30). We further observed a high colocalization between zDHHC5 and flotillin-1 by immunofluorescence (Fig. 2c). The zDHHC5 knockdown cells were subsequently used as a model to study the effect of endogenous flotillin-1 palmitoylation. As before, we performed CHX stability assays and found a decrease in flotillin-1 protein stability in the zDHHC5 knockdown compared to control cells (Fig. 2e). To further confirm that this degradation was through the proteosome, we performed rescue experiments with MG-132 treatment, which effectively restored flotillin-1 protein levels in the presence of CHX compared to vehicle control (Fig. 2f). Given that a 26S proteosome inhibitor (MG-132) was restoring protein expression in the zDHHC5 silenced cells, we questioned if blocking palmitoylation was increasing flotillin-1 poly-ubiquitination. To analyze the poly-ubiquitination state of flotillin-1, we utilized tandem ubiquitin binding entities (TUBE), a poly-ubiquitin resin conjugated to biotin, which allows for streptavidin enrichment and western blotting of the pulldown fraction which contains the poly-ubiquitylated proteins. Cell lysates from control or zDHHC5 knockdown cells, were incubated with or without the biotin conjugated TUBE and isolated by streptavidin enrichment (Fig. 2g). Western blotting of the enriched lysates revealed that zDHHC5 knockdown significantly increased the poly-ubiquitination of flotillin-1 (Fig. 2h), suggesting that zDHHC5 mediated flotillin-1 palmitoylation effects its stability by preventing its ubiquitin mediated proteasomal degradation.
3. Flotillin-1 palmitoylation defective TNBC cells display attenuated tumor growth and lung metastasis.
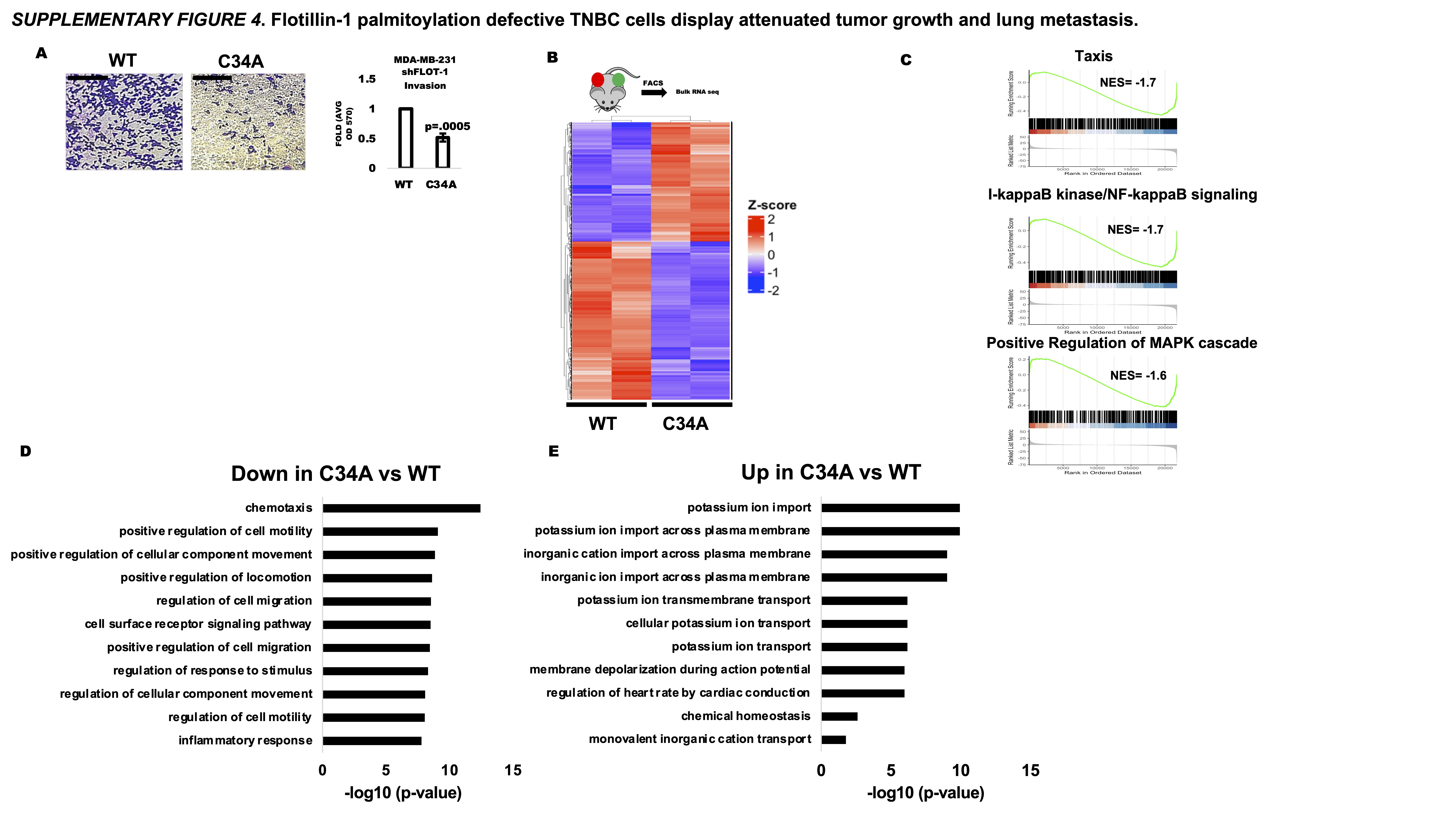
We initially observed in vitro that the flotillin-1 C34A expressing cells displayed decreased invasive capacities compared to flotillin-1 WT using collagen-I coated trans well membranes (Supplementary Fig. 4a) To test if these in vitro findings would translate into metastasis in vivo, we utilized our shF1 MDA-MB-231 cells stably re-expressed with flotillin-1 WT and C34A. Flotillin-1 WT and C34A cells were then counter labelled with lentiviral GFP and dTomato reporters, respectively, to track metastasis.
Flotillin-1-WT-GFP and flotillin-1-C34A-dTomato expressing MDA-MB-231 cells were subsequently implanted bilaterally into the left and right #4 mammary fat pad of SCID beige mice (Fig. 3 a,b). After 8 weeks of tumor growth, we analyzed both primary tumor size and lungs for metastasis (n=5 mice). At the end of the 8-week growth period, there was a significant reduction in primary tumor weight in the C34A vs WT tumors (Fig. 3c). We also assessed tumor proliferation and apoptosis by IHC staining for Ki67 and cleaved-caspase 3, respectively. IHC staining of the primary tumor sections revealed substantially less Ki67 staining in the C34A compared to WT (Fig. 3d). From the 5 mice, two WT and C34A flotillin-1 mice were selected for FACS sorting to isolate GFP+ and dTomato+ cells from these tumors for bulk RNA sequencing (Fig. 3e, Supplementary Fig. 4b). Differential expression analysis resulted in 1,118 upregulated and 570 downregulated transcripts between C34A vs WT flotillin-1 expressing tumors when filtered based on an absolute log2fold change of ±1 (Fig. 3e). Gene ontology analysis revealed a down-regulation of transcripts associated with cell motility, receptor signaling, and inflammatory responses while there was an up-regulation of transcripts involved in ion transport and membrane depolarization in C34A expressing cells vs WT (Fig. 3f., Supplementary Fig. 4. d,e). Gene set enrichment analysis also confirmed a negative enrichment in gene sets involved in cell taxis as well as NF-kb and MAPK signaling in the C34A flotillin-1 expressing tumors vs WT, in line with the gene ontology analysis (Supplementary Fig. 4c). We subsequently identified five transcripts from our differential expression analysis ( absolute log2fold change ± 1.5) that were also present in a 14 mRNA TNBC lung metastasis signature (31), including CXCL2, SELL, CD36, ICAM1, and G0S2. Collectively, these results demonstrate the ability of flotillin-1 palmitoylation inhibition to alter the migratory and metastatic cell phenotype at the transcript level (Fig. 3h). To assess if the transcriptional metastatic phenotype observed would translate to a functional phenotype, lungs from WT and C34A flotillin-1 tumor bearing mice (n=5) were analyzed for GFP+ and dTomato+ lung metastases. GFP and dTomato lung section immunostaining revealed multiple cases of multi-cellular GFP+ micro metastatic lung lesions with no observed dTomato+ (Fig 3 i,j). The absence of dTomato+ lung metastatic cells could be due to the drastically smaller primary tumor size and consequently less available cells to seed into circulation rather than an attenuated metastatic capability.
To ensure that the observed loss in lung metastasis from the C34A flotillin-1 expressing tumors was not merely due to a smaller number of cells initializing the metastatic cascade, we utilized an experimental lung metastasis model which removes the factor of primary tumor size and focuses on later stages of metastasis after cells have begun hematogenous dissemination. In this model, an equal number (5x105) of flotillin-1 WT and C34A luciferase expressing MDA-MB-231 cells were intravenously injected into the lateral tail vein of SCID beige mice (WT n=3, C34A n=4). Photon flux from the bioluminescence (BLI) imaging revealed a significant reduction in lung metastasis from the flotillin-1 C34A cells compared to WT (Fig. 3 k, l). Lung metastasis was further confirmed by performing H&E staining of lung sections (Fig. 3k). The current results demonstrate flotillin-1 palmitoylation to be an essential biological process for tumor progression and successful establishment of lung metastasis in a TNBC xenograft model.
4. Targeting flotillin-1 palmitoylation with a competitive peptide attenuates tumor growth and lung metastasis.
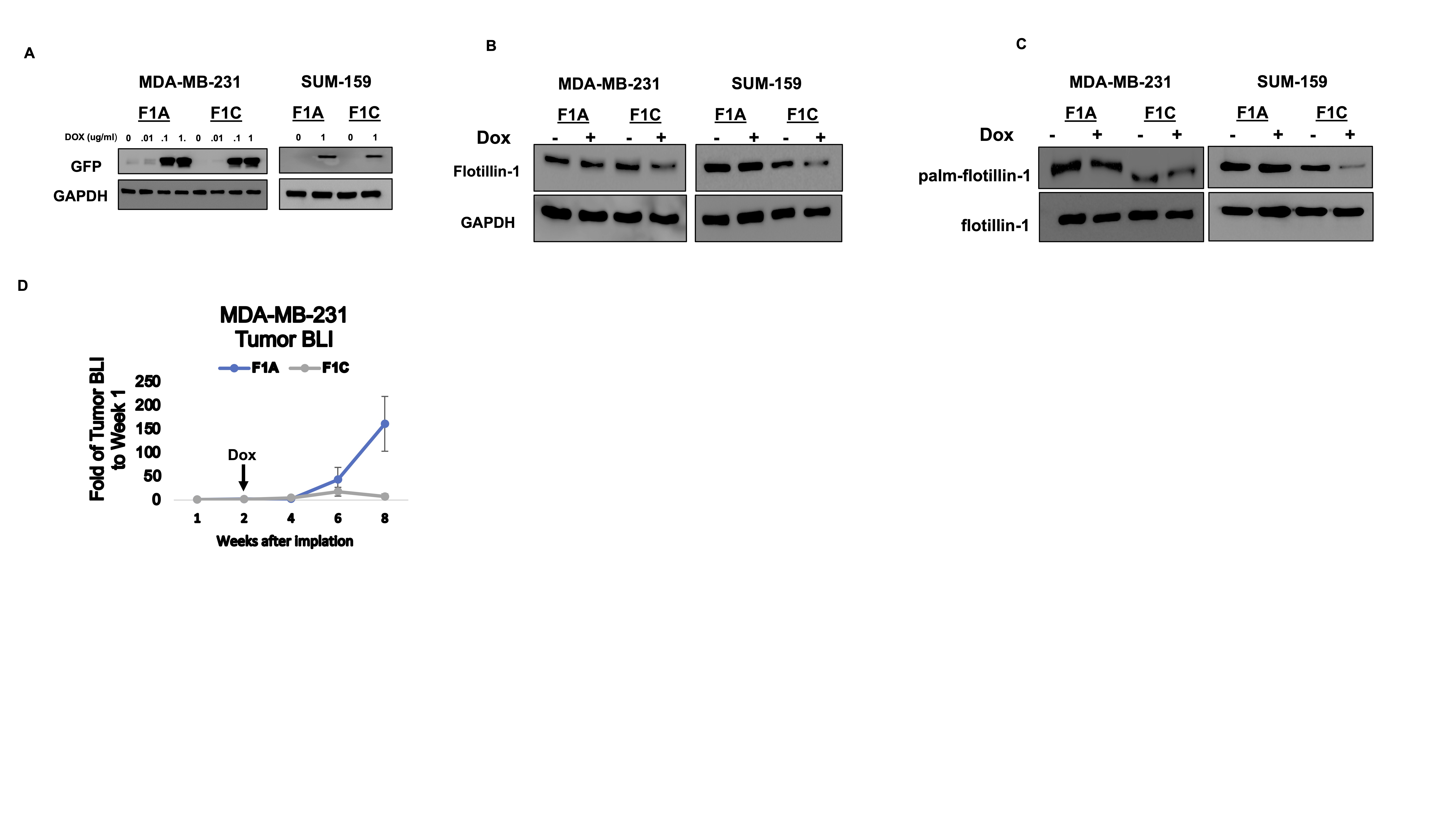
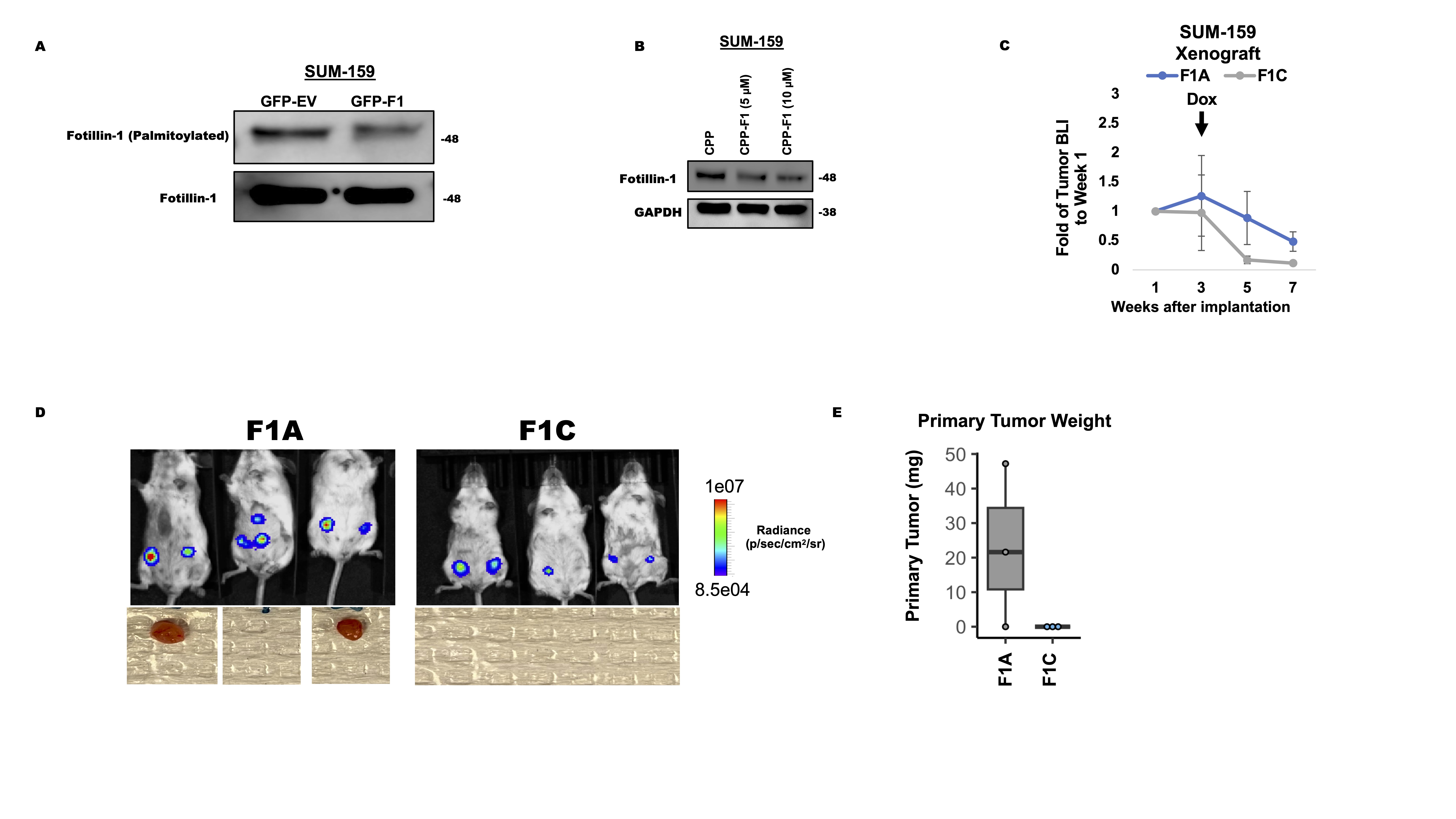
We have demonstrated that altering the palmitoylation status of flotillin-1 results in its degradation and attenuated tumor progression in vivo. In order to assess the ability to target this process for potential therapeutic benefit, we designed a competitive targeting peptide containing the palmitoylation sequence of flotillin-1, a strategy previously employed to block the palmitoylation of other proteins in preclinical models of solid tumors (29, 32). To study the effect of a palmitoylation blocking peptide in vitro, the coding sequence corresponding to flotillin-1 amino acid sequence (23-42) was cloned into a eGFP lentiviral vector. We subsequently transduced MDA-MB-231 cells with either an empty eGFP vector (GFP-EV) for a control or the flotillin-1 peptide sequence (23-42) fused to GFP (GFP-F1). For the peptide to successfully block endogenous flotillin-1 palmitoylation, it must first become palmitoylated, presumably through zDHHC5, to saturate the palmitate that is destined for flotillin-1 modification. We subsequently performed click chemistry labeling in MDA-MB-231 cells alone, expressing an empty GFP vector (GFP-EV), or expressing the flotillin-1 peptide fused to GFP (GFP-F1) to assess the ability of the flotillin-1 peptide to become palmitoylated. Click labeling revealed high levels of palmitoylation in the GFP-F1 peptide compared to that of GFP alone (Fig. 4a). To determine if the GFP-F1 peptide palmitoylation was through zDHHC5, we expressed the GFP-F1 peptide in shControl or shzDHHC5 cells followed by click chemistry labeling. GFP pulldown of the labelled proteins revealed a drastic reduction in GFP-F1 palmitoylation in the zDHHC5 depleted cells compared to control (Fig. 4b). Importantly, we found the zDHHC5 induced palmitoylation of the GFP-F1 peptide to substantially reduce the palmitoylation of endogenous flotillin-1 ostensibly by acting as a zDHHC5 palmitoylation decoy (Fig. 4d, Supplementary Fig. 6a). To study the ability to block flotillin-1 palmitoylation in a more therapeutically relevant manor, we fused the flotillin-1 palmitoylation targeting peptide sequence to a CPPtat , a well-known peptide sequence that encourages cellular uptake of therapeutics into cells (33) (Fig. 4e). In vitro treatment with the flotillin-1 peptide (CPP-F1) successfully inhibited flotillin-1 palmitoylation in MDA-MB-231 cells, without altering the palmitoylation state of two other zDHHC5 substrates, EZH2 and flotillin-2 (Fig. 4f). We further observed that this peptide decreased total flotillin-1 protein expression in both MDA-MB-231 and SUM-159 cells, providing in vitro evidence of successful flotillin-1 protein targeting through the delivery of a palmitoylation inhibiting peptide (Fig. 4g, Supplementary Fig. 6b). Knowing that the flotillin-1 peptide sequence successfully targets endogenous flotillin-1 protein palmitoylation, we questioned if this would provide any benefit in TNBC tumor progression in vivo. For this analysis, we utilized the delivery of the peptide through a lentiviral vector rather than fused to a CPPtat sequence to ensure that the peptide would be successfully delivered into the cells. The doxycycline inducible system was further utilized to provide temporal control of the flotillin-1 peptide expression. To generate these inducible flotillin-1 palmitoylation targeting constructs, we subcloned the flotillin-1 coding sequence corresponding to its amino acids (23-42) into tetracycline inducible lentiviral vector containing an eGFP to generate a GFP- flotillin-1 targeting peptide fusion, which was termed F1C (Fig 4g). We also subcloned a mutated (C34A) flotillin-1 peptide sequence into the same backbone vector as a control, further referred to as F1A (Fig. 4g). There was no observed GFP-peptide expression in the absence of doxycycline, suggesting no promoter leakiness (Supplementary Fig. 5a). Subsequent in vitro analysis revealed a reduction in flotillin-1 protein and palmitoylation status upon doxycycline addition in the F1C vs F1A cells (Supplementary Fig. 5b-c.) To assess the ability to target flotillin-1 protein in vivo through the delivery of a competitive peptide, MDA-MB-231 TNBC cells engineered to express either F1C or F1A constructs as well as a bicistronic dual mCherry-luciferase reporter were implanted into the #4 mammary fat pads of SCID beige mice to induce tumors. Upon palpable tumor detection, mice were switched to a diet containing 200 mg/kg of doxycycline to activate the expression of the flotillin-1 targeted (F1C) or control (F1A) peptides in the xenografted tumors. BLI detection of the primary tumors showed no change in tumor growth prior to the addition of doxycycline (Supplementary Fig. 5d), however, after 8-weeks of growth, there was a significant reduction in primary tumor weight (n=3) in the F1C vs F1A mice (Fig. 4 h,i, Supplementary Fig. 5d). Primary tumors were subsequently analyzed to detect levels of flotillin-1 palmitoylation, by acyl-biotin exchange (experimental details in materials and methods), which revealed substantial flotillin-1 palmitoylation inhibition in F1C tumors compared to F1A. It was further confirmed that F1C targeting peptide significantly decreased flotillin-1 total protein expression in primary tumors (n=3) compared to that of F1A control (Fig. 4l). To assess if targeting flotillin-1 palmitoylation and protein in the primary tumor would result in decreased metastasis, we performed ex vivo BLI detection in the lungs of tumor bearing mice. Unfortunately, we found that only two out of three lungs from the F1A mice contained metastasis large enough to be detected by BLI (Fig. 4h). As an alternative, we utilized the mCherry reporter that the cells also harbored to assess lung metastasis by FACS (Fig. 4j). When single cell suspensions from the lungs of tumor bearing mice were subjected to FACS analysis for mCherry expression, there was a significant reduction in percentage of mCherry+ cancer cells in the lungs of F1C vs F1A mice (n=3) (Fig 4j). We repeated this experiment using SUM-159 xenografted tumors, which also did not display altered tumor growth prior to doxycycline administration (Supplementary Fig. 6c). Surprisingly, there was an observed tumor regression post-doxycycline administration in both F1A and F1C mice that lead to an early termination point at week 7 instead of week 8. Regardless, we observed a pronounced decrease in tumor BLI in F1C vs F1A tumors (Supplementary Fig. 6 c,d). Shockingly, we did not observe any tumors (0 out of 3 mice) in the F1C mice compared to F1A, which contained established tumors 2 out of 3 mice (Supplementary Fig. 6 c,d,f). Given the ability of the expressed peptide from a lentiviral vector to target flotillin-1 protein in vivo, we questioned if it could be delivered in the same format we utilized in vitro. To this end, we dissolved the flotillin-1 targeting CPP-F1 peptide in water and administered it by I.P. injection to tumor bearing mice along with water only injections as a control. 24 hours post injection, tumors were analyzed for total flotillin-1 protein. Treatment with 150ug of CPP-F1 peptide substantially reduced flotillin-1 protein in tumors compared to water only controls (n=2) (Fig. 4m). Collectively, these results demonstrate the ability to target flotillin-1 protein in vivo through delivery of a competitive peptide and provide proof-of-concept data demonstrating flotillin-1 targeted peptide delivery as an effective strategy to promote its degradation and subsequently impede tumor growth and metastasis in xenograft models of TNBC.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}