5.1 Materials
Peptide Synthesis: Fmoc-protected amino acids were purchased from ORPEGEN (Heidelberg, Germany). Fmoc-D-homocysteine(Trt)-OH and Fmoc-NH-PEG(4)-OH were purchased from Iris Biotech (Marktredwitz, Germany). Peptide resins, 1-hydroxy benzotriazole (HOBt), ethanedithiol (EDT), diethyl ether, and trifluoracetic acid (TFA) were obtained from Merck (Darmstadt, Germany). N,N’-diisopropyl carbodiimide (DIC) and 2-cyano-2-(hydroxyimino) acetic acid ethyl ester (oxyma) were purchased from Iris Biotech (Marktredwitz, Germany). Dimethylformamide (DMF) and dichloromethane (DCM) were purchased from Biosolve (Valkenswaard, Netherlands), acetonitrile (ACN) was obtained from VWR (Darmstadt, Germany). O-(7-Azabenzotriazol-1-yl)-N,N,N,N-tetramethyluronium hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), piperidine, and thioanisole (TA) were obtained from Sigma-Aldrich (St. Louis, USA). 6-Carboxytetramethylrhodamine (Tam) was purchased from emp biotech (Berlin, Germany).
Cell Culture: Cell culture media (Dulbecco’s Modified Eagle’s Medium (DMEM), Ham’s F12), as well as trypsin-EDTA, Dulbecco’s Phosphate-Buffered Saline (DPBS), and Hank’s Balanced Salt Solution (HBSS), were obtained from Lonza (Basel, Switzerland). Fetal bovine serum (FBS) was from Biochrom GmbH (Berlin, Germany). Hygromycin B was purchased from Invivogen (Toulouse, France), and Opti-MEM was obtained from Life Technologies (Basel, Switzerland). LipofectamineTM 2000 was obtained from Invitrogen (Carlsbad, CA, USA). MetafecteneProTM was received from Biontex Laboratories GmbH (München, Germany). Coelenterazine H was purchased from DiscoverX (Fremont, CA, USA), Hoechst33342 nuclear stain was obtained from Sigma-Aldrich (St. Louis, MO, USA). Bovine arrestin-3 was fused to Rluc8 and cloned into pcDNA3 vector for BRET studies. Primers for PCR were bought from Biomers (Ulm, Germany). Furimazine was purchased from Promega (Madison, WI, USA).
5.2 Peptide Synthesis
All peptides were synthesized by 9-fluorenylmethoxycarbonyl/tert-butyl (Fmoc/tBu) solid-phase peptide synthesis strategy on a scale of 15 µmol on a Wang resin preloaded with the first amino acid, or on a 2-chlorotrityl chloride resin. All reactions were performed at rt unless stated otherwise. All standard amino acids were coupled using a Syro II peptide synthesizer (MultiSynTech, Bochum, Germany). Coupling reactions were performed twice with 8 equiv of the respective, Fmoc-protected amino acid activated in situ with equimolar amounts of oxyma and DIC in DMF for 30 min. Fmoc-removal was achieved by reaction with 40 % (v/v) piperidine in DMF for 3 min and 20% (v/v) piperidine in DMF for 10 min, the resin was washed with DMF and DCM after every reaction. Fmoc-D-hCys(Trt)-OH and Fmoc-PEG(4)-OH were manually coupled to the peptide by reaction with 5 equiv and equimolar amounts of HOBt and DIC in DMF overnight. N-terminal Tam-labeling was achieved by reaction with 2 equiv Tam, 1.9 equiv HATU, 2 equiv DIPEA in DMF for 2 h.
[N-C]-c(chemerin-9) was synthesized on a 2-chlorotrityl chloride resin, the first amino acid was coupled to the resin by reaction with 1.5 equiv Fmoc-Ser(tBu)-OH, 6 equiv DIPEA in DCM overnight. All following amino acids were coupled as described above. After protected cleavage from the resin with 10% (v/v) acetic acid, 10% (v/v) trifluoroethanol in DCM for 2 h at rt, a lactam bond between the N- and C-terminus was formed by incubation with 5 equiv HOBt, DIC in DCM for 72 h at rt.
All peptides were cleaved by reaction with TFA/EDT/TA (90:3:7, v:v:v) for 3 h and precipitated in ice-cold diethyl ether/hexane (1:3). After full cleavage, the crude peptides were precipitated from cold diethyl ether/hexane at -20°C for at least 3 h, washed with diethyl ether and collected by centrifugation. The thiol-containing peptide was dissolved in refolding buffer (20% ACN, 0.15 M NaCl, 25 mM Tris, pH 7.7) and incubated at rt for 72 h for cyclization to yield the disulfide containing peptide [4-9]-c(chemerin-9). All peptides were purified by RP-HPLC on a Kinetex 5 µm XB-C18 100 Å column (Phenomenex, Torrence, USA), purity and identity were confirmed by RP-HPLC on a Jupiter 4 µm Proteo 90 Å C12 (Phenomenex), MALDI-ToF MS on an Ultraflex II and ESI MS on an HCT ESI (Bruker Daltonics, Billerica, USA). RP-HPLC was performed employing linear gradients of eluent B (0.08% TFA in ACN) in eluent A (0.1% TFA in H2O).
5.3 Protein Expression
Full-length chemerinS157 was produced as a His10-fusion protein in E.coli BL21 (DE3) as described previously [51]. In brief, the plasmid DNA (His10-chemerinS157 in pET16b) was transformed into E.coli. Bacteria were grown in LB medium supplemented with 0.1 mg/mL ampicillin. Upon reaching OD600 = 0.8, expression was induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG). After 6 h expression at 37° C, cells were harvested by centrifugation. Resuspended in base buffer (0.5 M NaCl, 25 mM Tris/HCl, pH 7.8), the cells were lysed using a FastPrep-24 bead beating lysis system (MP Biomedicals, Irvine, USA). After washing with base buffer supplemented with 2 M urea, inclusion bodies were solubilized in base buffer containing 8 M urea. The solubilized protein was purified by immobilized metal affinity chromatography and refolded by stepwise dialysis employing decreasing urea concentrations and a cysteine/cystamine redox pair. Purity and identity were confirmed by SDS-PAGE, RP-HPLC, and MALDI ToF MS (Ultraflex III, Bruker).
5.4 Mutagenesis
All mutations were introduced into an hGPR1-eYFP pVitro2 plasmid kindly provided by Stefan Schultz [51]. All PCR reactions were carried out using Phusion polymerase. The SigP-Cys-E3-hGPR1-eYGP construct was cloned by PCR utilizing the primers shown in Table 2 by introducing the Cys-E3 tag and the signal peptide in two sequential PCR reactions. The Nluc-GPR1-eYFP construct was cloned by overlap extension PCR using the primers display in Table 2, exploiting the MluI and XbaI restriction sites. The secNluc-pNL1.3 vector was purchased from Promega. Point mutations were introduced into the Nluc-GPR1-eYFP construct using the QuickChange mutagenesis protocol with primers carrying the respective mutations. The success of any mutagenesis or cloning reactions was verified by Sanger sequencing.
Table 2: Primers used in PCR reactions to introduce either the E3 tag and the signal peptide for increased membrane expression, or the N-terminal Nluc for BRET based binding assays. Parts in bold correspond to the sequences not present in the respective template.
|
Primer
|
Sequence
|
|
E3-tag forward
|
ATGTGCGAGATCGCCGCCCTGGAGAAGGAGATCGCCGCCCTGGAGAAGGAGATCGCCGCCCTGGAGAAGGGCGGCTCAATGGAAGATTTGGAGGAAACATTATTTG
|
|
E3-tag reverse
|
GCGATCTCGCACATGGTGGCACGCGTGG
|
|
SigP forward
|
ATGCAGCCGCCTCCAAGTCTGTGCGGACGCGCCCTGGTTGCGCTGGTTCTTGCCTGCGGCCTGTCGCGGATCTGGGGATGCGAGATCGCCGCCCTGGAG
|
|
SigP reverse
|
GAGGCGGCTGCATGGTGGCACGCGTGG
|
|
Nluc-forward
|
ATATACGCGTGCCACCATGAACTCCTTCTCCACAAGCGCC
|
|
Nluc-linker-reverse
|
GCTGCCTCCGCCTCCGCTCGCCAGAATGC
|
|
linker-GPR1-forward
|
GGAGGCGGAGGCAGCGAAGATTTGGAGGAAACATTATTTGAAG
|
|
GPR1-eYFP-reverse
|
ATATTCTAGACTACTTGTACAGCTCGTCCATGCCGAG
|
5.5 Cell Culture
HEK293 and COS-7 cells were cultivated in DMEM/Ham’s F12 supplemented with 15% FBS or DMEM supplemented with 10% FBS, respectively. HEK293 and COS-7 cells were authenticated by DNA barcoding in 2017. HEK293 cells stably transfected with GPR1-eYFP were cultivated in DMEM/Ham’s F12, 15% FBS supplemented with 100 µg/mL hygromycin. All cells were cultivated in T75 cell culture flasks at 37°C, 95% humidity, 5% CO2. All experiments with eukaryotic cells were carried out at 37° C unless stated otherwise.
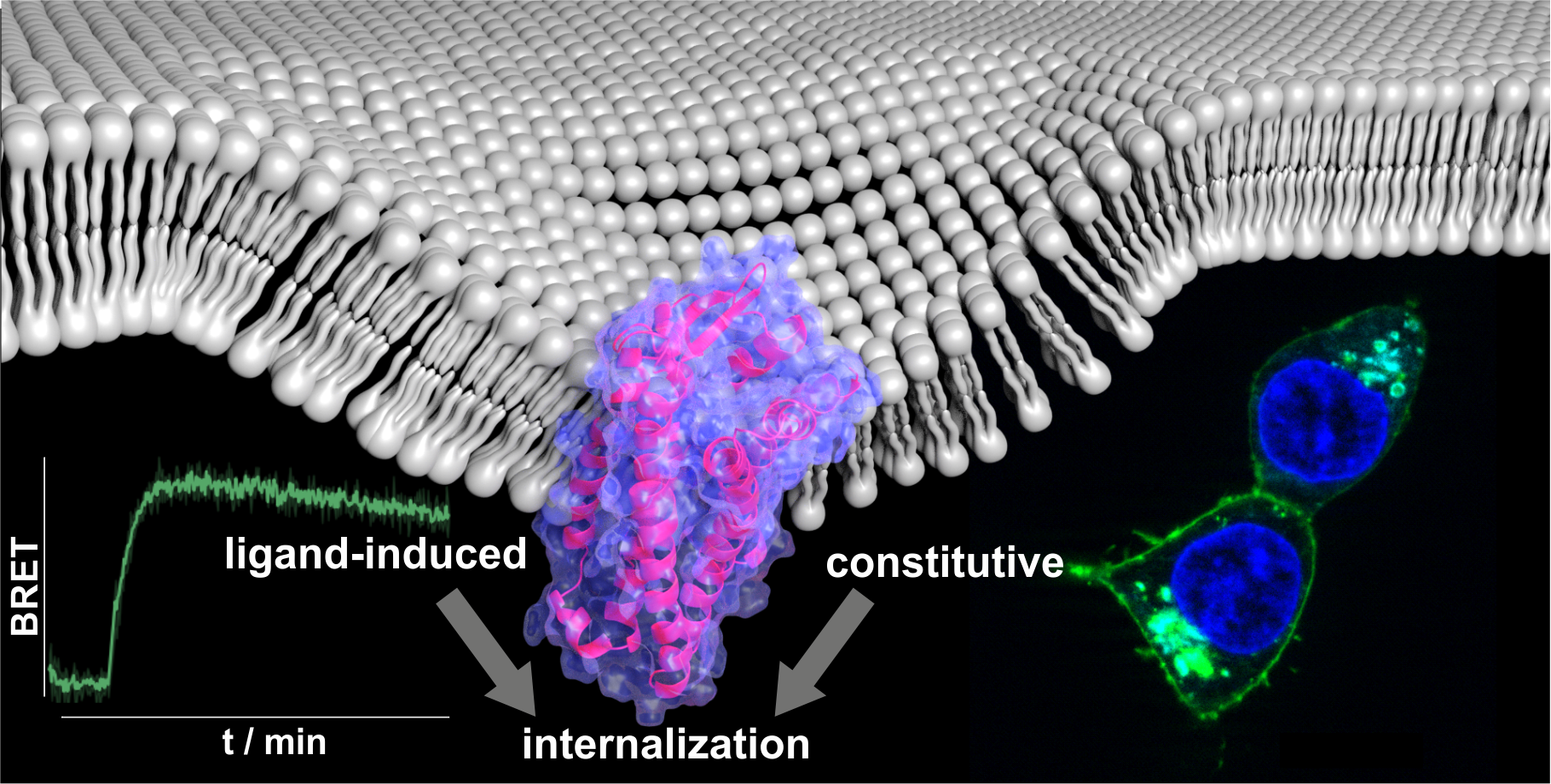
5.6 Characterization of Arrestin Recruitment
Arrestin recruitment was characterized by measuring the bioluminescence resonance energy transfer (BRET) ratio between luciferase-tagged arrestin3 and either GPR1-eYFP or CMKLR1-eYFP as described previously [52]. In brief, COS-7 cells in 75 cm2 cell culture flasks were transiently transfected overnight with 7800 ng GPR1-eYFP or CMKLR1-eYFP in pVitro2 and 200 ng Rluc8-Arr3 in pcDNA3 using MetafectenePro according to the manufacturer’s protocol. One day post-transfection, cells were detached using trypsin/EDTA, resuspended in 20 mL DMEM/Ham’s F12 w/o phenol red supplemented with 15 % FBS and seeded in white 96 well plates (100 µL cell suspension/well) and grown overnight. Prior to the assay, the medium was replaced by 100 µL BRET buffer (25 mM HEPES, pH 7.4 in HBSS), and 50 µL of the Renilla luciferase substrate Coelenterazine H (Nanolights) was added (final concentration 4.2 µM), followed by 5 min incubation. For kinetic measurements, cells were stimulated with agonist in buffer or buffer alone, and fluorescence was measured for 20 min using a Tecan Infinite M 200 Reader (Tecan, Männerdorf, Switzerland) using the filter sets Green1 (YFP emission, luminescence 520-570 nm) and Blue1 (luciferase luminescence 370-480 nm). For concentration-response curves, cells were stimulated with agonist or blank, and luminescence was measured after 10 min. BRET signal was calculated as the ratio of fluorescence divided by luminescence, netBRET signal was calculated by subtracting the BRET signal of unstimulated wells from the respective samples.
5.7 Ligand BRET
Ligand binding was characterized using a NanoBRET approach with Tam-labeled ligands and hGPR1-eYFP N-terminally modified with a NanoLuc [53]. COS-7 cells in 25 cm2 cell culture flasks were transfected with 4000 ng of the respective Nluc-GPR1-eYFP construct using MetafectenePro according to the manufacturer’s protocol. One day post-transfection, cells were detached using trypsin/EDTA, resuspended in 10 mL phenol red-free DMEM/Ham’s F12, 15% FBS, seeded into solid-black 96 well plates (100 µL/well), and grown overnight. Before the assay, the medium was replaced by 100 µL BRET buffer. Cells were stimulated with Tam-labeled peptides, and 50 µL of furimazine in BRET buffer was added. Measurements were performed using a Tecan Spark plate reader (Tecan, Männerdorf, Switzerland), measuring Tam-emission (550-700 nm), and NanoLuc luminescence (430-470 nm). BRET and netBRET signals were calculated as described above.
5.8 Live-Cell Fluorescence Microscopy
For fluorescence microscopy, HEK293 cells were seeded into 8 well 15µ-slides (Ibidi, 140,000 cells/200 µL/well) coated with poly D-lysine and grown overnight. Next, cells were transfected with 900 ng of the respective GPR1-eYFP plasmid and, where applicable, 100 ng of either rab4-CFP, rab11-CFP, or mCherry-arrestin3. Transfection was achieved using Lipofectamine 2000 according to the manufacturer’s protocol. One day post-transfection, fluorescence microscopy experiments were performed on an AxioVision Observer.Z1 microscope equipped with an ApoTome imaging system (Zeiss, Jena, Germany). Before the experiment, cells were starved in OptiMEM reduced serum medium containing Hoechst 33342 for 30 min. To observe arrestin recruitment, cells were stimulated with 1 µM chemerin-9 in OptiMEM for the indicated period. To observe peptide uptake, cells were stimulated with 1 µM Tam-chemerin-9 in OptiMEM, which was replaced with acidic wash (50 mM glycine, 100 mM NaCl, pH 3) in HBSS after the indicated time, followed by two washing steps with OptiMEM. Microscopy was carried out in OptiMEM; the exposure time was held constant whenever changes over time were observed.
5.9 Peptide-Templated On-Surface Labeling
Peptide-templated acyl transfer for selective labeling of membrane receptors was carried out as described previously [30]. Because E3-tagged GPR1 was not expressed in the membrane, the endothelin B receptor N-terminal signal peptide was attached. This signal peptide improves transport to the membrane and is cleaved upon successful membrane integration [54]. HEK293 cells were seeded out and transfected with SigP-Cys-E3-GPR1-eYFP in pVitro2 as described above and incubated in Hoechst 33342 in OptiMEM for 30 min, followed by 10 min incubation in 20 mM HEPES in HBSS, pH 7 (labeling buffer). Labeling of cell surface receptors was achieved by 5 min incubation with 150 nM Tam-K3 peptide probe in labeling buffer supplemented with 0.1 mM TCEP. The cells were washed by incubation with 200 mM NaHCO3 in DPBS w/o Ca2+ and Mg2+, pH 3 for 1.5 min, followed by two washing steps with labeling buffer. Microscopy studies were finally carried out in labeling buffer as described above. To prevent receptor internalization, the cells were cooled on ice, followed by labeling and washing with ice-cold solutions and microscopy at rt. The Tam-K3 peptide was synthesized as described before [31]. The fraction of Tam-fluorescence in the membrane was determined by quantifying total and intracellular fluorescence for each cell individually in imageJ. The fluorescence fraction in the membrane was defined as the difference between intracellular and total fluorescence divided by total fluorescence.
5.10 Peptide Uptake using a High Content Imaging System
To quantify receptor-mediated peptide uptake, the intracellular accumulation of Tam-fluorescence was observed at different time points. HEK293 cells stably transfected with GPR1-eYFP were seeded into µclear, black 96 well plates (100,000 cells/well) coated with poly D-lysine and grown overnight. Before the experiment, cells were incubated with Hoechst 33342 in OptiMEM, which was replaced with pure OptiMEM after 30 min. Cells were stimulated with 1 or 10 µM of the respective, Tam-labeled peptide for the specified periods, followed by washing with acidic wash (50 mM glycine, 100 mM NaCl, pH 3) in HBSS. Microscopy was carried out in OptiMEM using an ImageXpress Micro Confocal High-Content Imaging System (Molecular Devices, San José, United States), using the appropriate filters for the respective fluorophores. The fluorescence intensity per cell was automatically analyzed for each well by a module detecting the nuclei (5-30 µm in diameter and 100 gray levels above background) and the granules by Tam-peptide fluorescence (2-5 µm in diameter, 70 gray levels above background).
5.11 Statistical Analysis
Linear and nonlinear regression, statistical analysis, and calculation of mean, SEM or SD was carried out using GraphPad Prism 8 except for BRET kinetics, where mean ± 95% CI values were plotted using seaborn and matplotlib in python 3. Linear regression of ΔΔG vs log(EC50) values was calculated in python 3 using the scipy stats module. The applied statistical tests, including sample sizes, are given in the respective figure legends.
5.12 Homology Modeling
Homology models of GPR1 in the apo state were produced using RosettaCM [55] with the crystal structures of C5aR1 [56] (PDB: 6c1r), CCR9 [57] (PDB: 5lwe), CXCR4 [58] (PDB: 3odu), APJR [59] (PDB: 5vbl) and AT1R [60] (PDB: 4zud) as templates as described previously [27, 28]. In brief, crystal structures were stripped of fusion proteins, ions, etc., followed by threading of the CMKLR1 sequence onto the template structures according to the sequence alignment given in Table S1. The threaded templates were hybridized giving a chimeric template, which was subjected to a Monte Carlo-based energy minimazion. Rosetta 3.9 was used for all modeling steps, 1500 homology models were produced in total.
5.13 Peptide Docking
To include structurally diverse templates for peptide docking, the homology models were clustered based on Cα RMSD, and chemerin-9 was docked into the ten best scoring models by total score from each of the three largest clusters using Rosetta FlexPepDock ab initio. [61] One sided restraints for the identified binding residues were applied to keep the peptide in the binding pocket, and a loop conformation of the peptide was enforced by a distance restraint between the peptide N- and C-terminus. An interaction between chemerin-9 residue F8 and CMKLR1 residues V4.67 and F4.69 was enforced by a distance restraint. In total, 25,000 models were produced. After clustering, the best scoring models from the cluster that best represented the experimental data were energy minimized using Rosetta FastRelax. The 20 best scoring final models by interface score ΔG separate were analyzed using a per residue energy breakdown. A detailed description of the modeling and docking process is given in the Supplementary Information.
{kind=link}