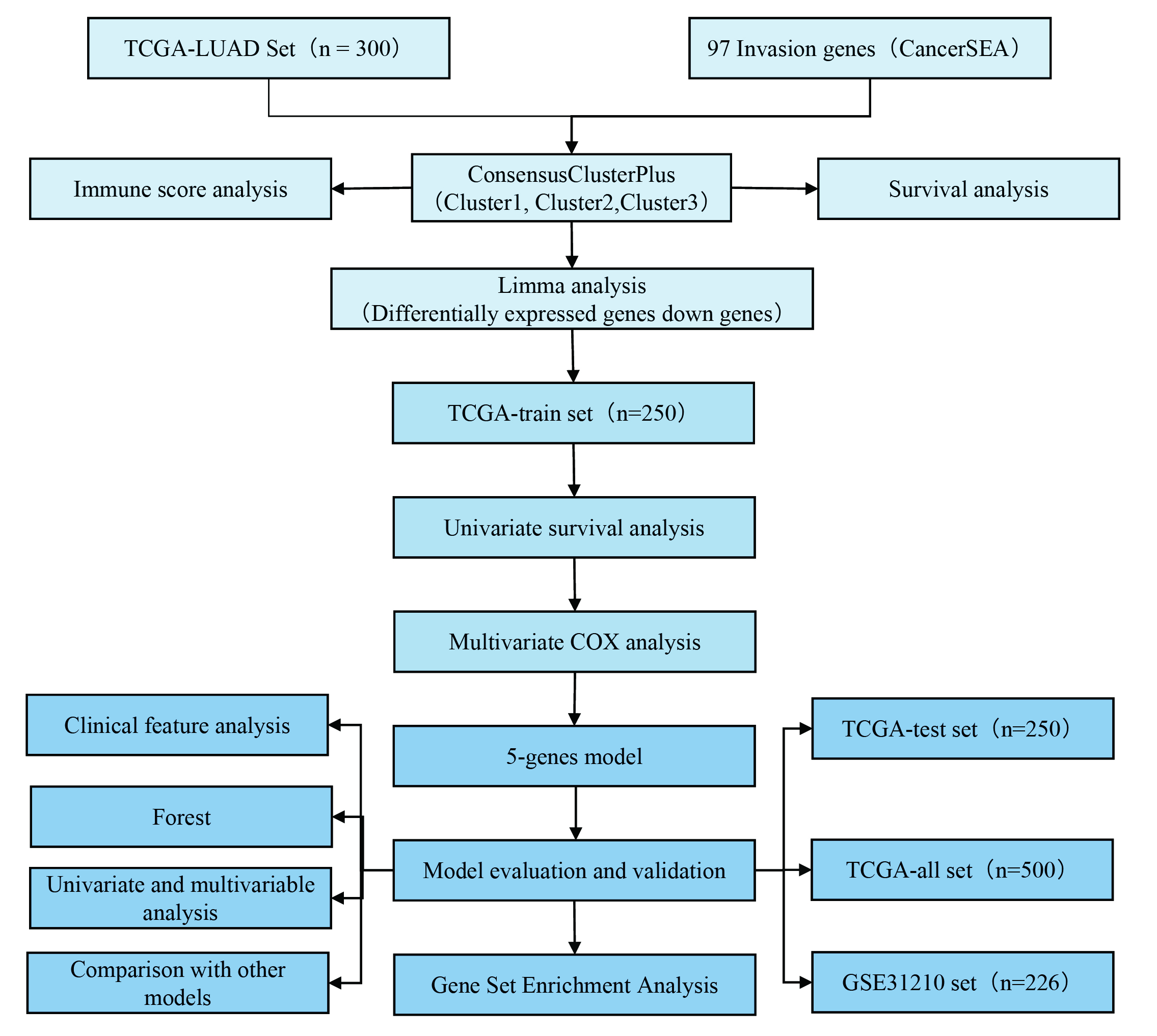
Study flow chart
To make the research easier for readers to understand, we drew a methodology flow chart (Supplementary Fig. 1).
Molecular typing of LUAD based on invasion-related genes
Through univariate Cox analysis of the 97 invasion-related genes in the TCGA expression profile, 19 genes were found to be associated with LUAD prognosis (p < 0.01; Supplementary Table 3). Consistent cluster analysis showed that the samples could be clustered at k = 3 (Fig. 1A). The expression levels of the invasion-related genes in the 3 subtypes are shown in Fig. 1B. These levels were different among the C1, C2, and C3 subtypes. Most of the genes were highly expressed in the C3 subtype and lowly expressed in the C2 subtype. We further analyzed the relationships between the 3 subtypes and prognosis. The results showed that there were significant differences between the 3 subtypes. The prognoses of patients with the C2 subtype were the best, and those of patients with the C3 subtype were the worst (log-rank p < 0.05; Fig. 1C, D).
Identification and functional analysis of DEGs among subtypes
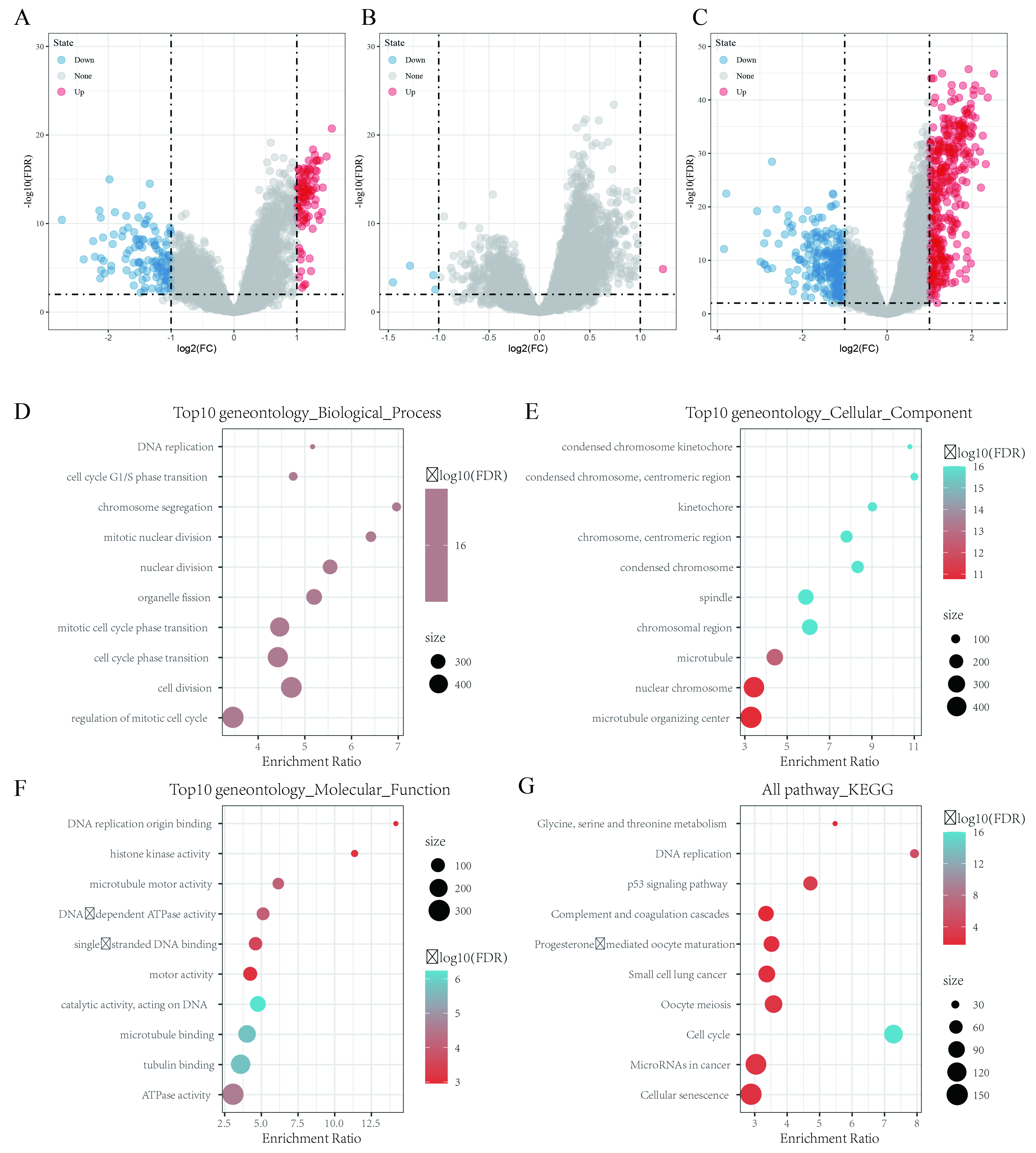
The DEGs between C1, C2 and C3 were identified by using the limma package in R. The volcano map of the DEGs between C1 and C3 is shown in Supplementary Fig. 2A; there were 98 upregulated genes and 123 downregulated genes. The volcano map of the DEGs between C1 and C2 is shown in Supplementary Fig. 2B; there was 1 upregulated gene and 4 downregulated genes. The volcano map of the DEGs between C2 and C3 is shown in Supplementary Fig. 2C; there were 389 upregulated genes and 267 downregulated genes.
A total of 669 DEGs between C1/C2, C2/C3, and C1/C3 were obtained, and these DEGs were further analyzed by KEGG pathway and GO functional enrichment analyses using the WebGestaltR (V0.4.2) software package in R. The biological functions of the top 10 genes enriched in biological processes (Supplementary Fig. 2D), cellular components (Supplementary Fig. 2E), and molecular functions (Supplementary Fig. 2F) were visualized. The KEGG pathway analysis results showed that the DEGs were significantly enriched in the cell cycle, DNA replication, p53 signaling pathway, microRNAs in cancer, small cell lung cancer, and other tumor-related pathways (Supplementary Fig. 2G).
Clinical correlations of molecular subtypes and comparison with existing subtypes
The distributions of different clinical features among the C1, C2, and C3 subtypes were compared. The results showed that there were significantly more C2 patients than C1 and C3 patients in the T1, N0, and Stage I samples, while there were significantly fewer C2 patients than C1 and C3 patients in the T2, N1, and Stage II samples (Fig. 1E-G). The number of survivors in the C2 group was significantly higher than in the C1 and C3 groups (Fig. 1H). These results confirmed that patients with the C2 subtype had the best prognoses.
Previous studies have analyzed 33 cancers in the TCGA database. These studies clustered non-blood tumors into 6 immune subtypes based on the distributions of various features, such as macrophages, immune-infiltrating lymphocytes, transforming growth factor-beta response, interferon-γ response, and wound healing; these subtypes include C1 (wound healing), C2 (INF-γ predominance), C3 (inflammation), C4 (lymphocyte depletion), C5 (immunological silencing), and C6 (transforming growth factor-beta predominance), among which C1 and C6 have been associated with poor prognosis(Thorsson et al.,, 2018). By comparing the molecular subtypes with these immune subtypes, it was found that most LUAD patients in the TCGA dataset belonged to the C1, C2, and C3 immune subtypes (about 89.5%), and there were no patients with the C5 immune subtype in the LUAD TCGA dataset (Fig. 1I). By comparing the distributions of the molecular and immune subtypes, it was found that patients with the C3 molecular subtype showed the highest proportion of the C2 immune subtype, reaching 54% (Fig. 1J). The proportion of the C2 immune subtype among the C2 molecular subtype was lower, and the proportion of the C3 immune subtype was higher than that of the C3 molecular subtype. The survival curve analysis results showed that there were significant differences in OS among the immune subtypes (p < 0.05; Fig. 1K). These results suggested that the prognosis of the C3 immune subtype was better than that of the C2 immune subtype.
Comparison of immune scores among subtypes
The relationships between the molecular subtypes of the TCGA dataset and immune scores were identified by using the ESTIMATE software package in R, MCPcounter, and the ssGSEA method in the GSVA package. The results showed that there were significant differences in immune scores among the different subtypes (Fig. 2A-C). The heat map of immune scores among the 3 subtypes is shown in Fig. 2D.
Construction of risk model
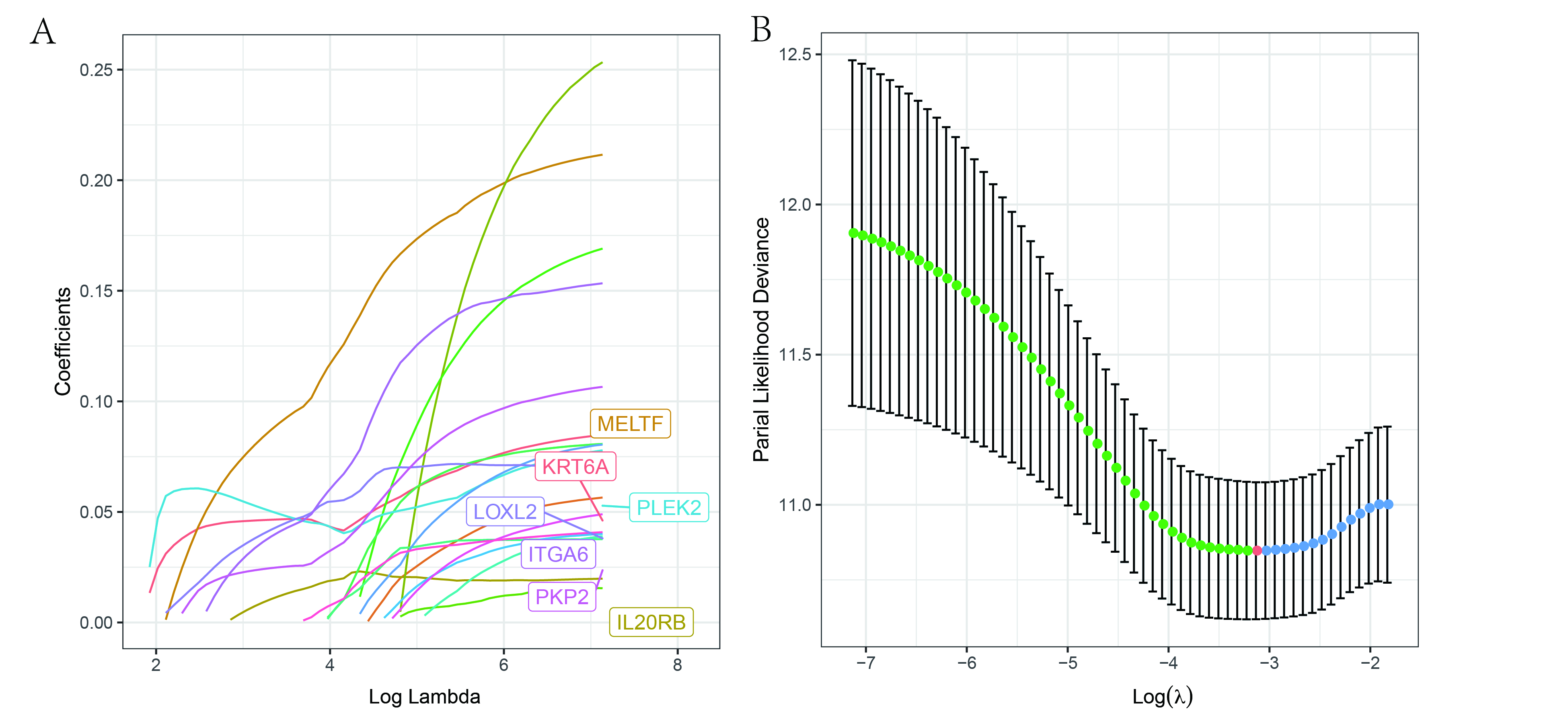
The 500 samples in the TCGA dataset were grouped according to the training set-to-validation set ratio of 1:1, and the univariate Cox proportional hazards regression model method was used to evaluate the 669 DEGs between the molecular subtypes. A total of 29 genes were found to be associated with prognosis (Supplementary Table 4). Lasso regression was used to further compress the 29 genes. The trajectory of each independent variable is shown in Supplementary Fig. 3A. As lambda decreased, the number of independent variable coefficients tending to 0 increased. We used 10-fold cross-validation to build the model and analyzed the confidence interval (CI) under each lambda (Supplementary Fig. 3B). When lambda equaled 0.003518527, the model was considered optimal, and 12 genes (KRT6A, MELTF, IL20RB, PLEK2, LOXL2, IRX5, SLC16A11, FAM189A2, ITGA6, PKP2, MS4A1, CRTAC1) were selected as target genes. The AIC algorithm was used to further compress these 12 target genes, and 5 target genes (KRT6A, MELTF, IRX5, MS4A1, CRTAC1) were finally obtained.
The Kaplan-Meier curves of the 5 genes are shown in Supplementary Fig. 3C-G. The 5 genes could be divided into 2 groups with high and low risk (p < 0.05). The final 5-gene signature formula was: RiskScore = 0.08073881 * KRT6A + 0.18237095 * MELTF − 0.17903164 * IRX5–0.26862737 * MS4A1–0.09946249 * CRTAC1.
Risk scores were further converted into Z-scores. Samples with scores > 0 were divided into the high-risk group, and samples with scores < 0 were divided into the low-risk group. A total of 119 samples were divided into the high-risk group, and 131 samples were divided into the low-risk group. The survival curve results showed that there was a significant difference in prognosis between the 2 groups (p < 0.0001; Fig. 3A).
The risk score distributions of the samples were calculated according to expression levels and then plotted (Fig. 3B). The survival times of the samples with high risk scores were significantly shorter than those of the samples with low risk scores, suggesting that samples with high risk scores had worse prognoses. The timeROC software package in R was used to analyze the prognostic classification efficiency of risk scores. The model had a large area under the curve (AUC) at 1, 3, and 5 years; the 1-year AUC was 0.72, and the 5-year AUC was 0.74 (Fig. 3C).
Verification of risk model robustness in internal and external datasets
The robustness of the model was verified by the internal dataset (TCGA validation set and all datasets) and external dataset (GSE31210 dataset). In all datasets, the same model and coefficients as those in the training set were used. The survival curve showed significant differences between the high- and low-risk groups in the verification set and all datasets (Fig. 3D and Fig. 3G).The risk score of each sample was calculated according to gene expression, risk score distributions were plotted in TCGA internal validation set and all datasets in Fig. 3E and Fig. 3H. The classification efficiencies of prognosis prediction at 1, 3, and 5 years in the TCGA testing cohort and entire TCGA cohort are shown in Fig. 3F and Fig. 3I, respectively. The 1-year AUC reached 0.73 in both datasets.
Z-score transformation of risk scores was performed in GSE31210 dataset. Samples with risk scores > 0 after Z-score transformation were divided into the high-risk group, and samples with risk scores < 0 after Z-score transformation were divided into the low-risk group. This resulted in 94 samples in the high-risk group and 132 samples in the low-risk group. The survival curve showed a significant difference between the high- and low-risk groups (p = 0.0028; Fig. 3J).
The risk score distribution of the samples in the GSE31210 cohort was consistent with that of the training set (Fig. 3K). Receiver operating characteristic (ROC) analysis showed that the 1-year AUC reached 0.79 (Fig. 3L).
Relationships among risk scores, clinical features, and molecular subtypes
Survival analysis of different clinical subgroups was carried out based on risk scores. The results showed that the 5-gene signature could significantly distinguish age, sex, tumor/node/metastasis (TNM) stage, stage, recurrence, chemotherapy, and smoking status (current smoker, never smoked, reformed smoker) samples into high- and low-risk groups (p < 0.05; Fig. 4A-T). The 5-gene signature could not divide the M1 samples into 2 groups with significant prognostic difference, which may be due to the small number of M1 samples (p > 0.05; Fig. 4J). In general, our model could be used as a prognostic marker for different clinical subgroups if the sample size was appropriate.
Risk score distributions in terms of different clinical features were also assessed. The results showed that there were no significant differences in terms of age or stage (p > 0.05; Fig. 5B, E). Risk scores showed significant differences in terms of sex (female, male), T stage (T1, T2, T3, T4), N stage (N0, N1, N2, N3), stage (Stage I, Stage II, Stage III, Stage IV), and smoking status (current smoker, never smoke, reformed smoker) (p < 0.05; Fig. 5A, C, D, F, G). We also compared risk scores among the 3 subtypes (C1, C2, C3). The results showed that the risk scores of C3 subtype samples with poor prognosis were significantly higher than those of C2 subtype samples with good prognosis (Fig. 5H), which further suggested that high risk scores were associated with poor survival outcomes.
Relationships between risk scores and pathways
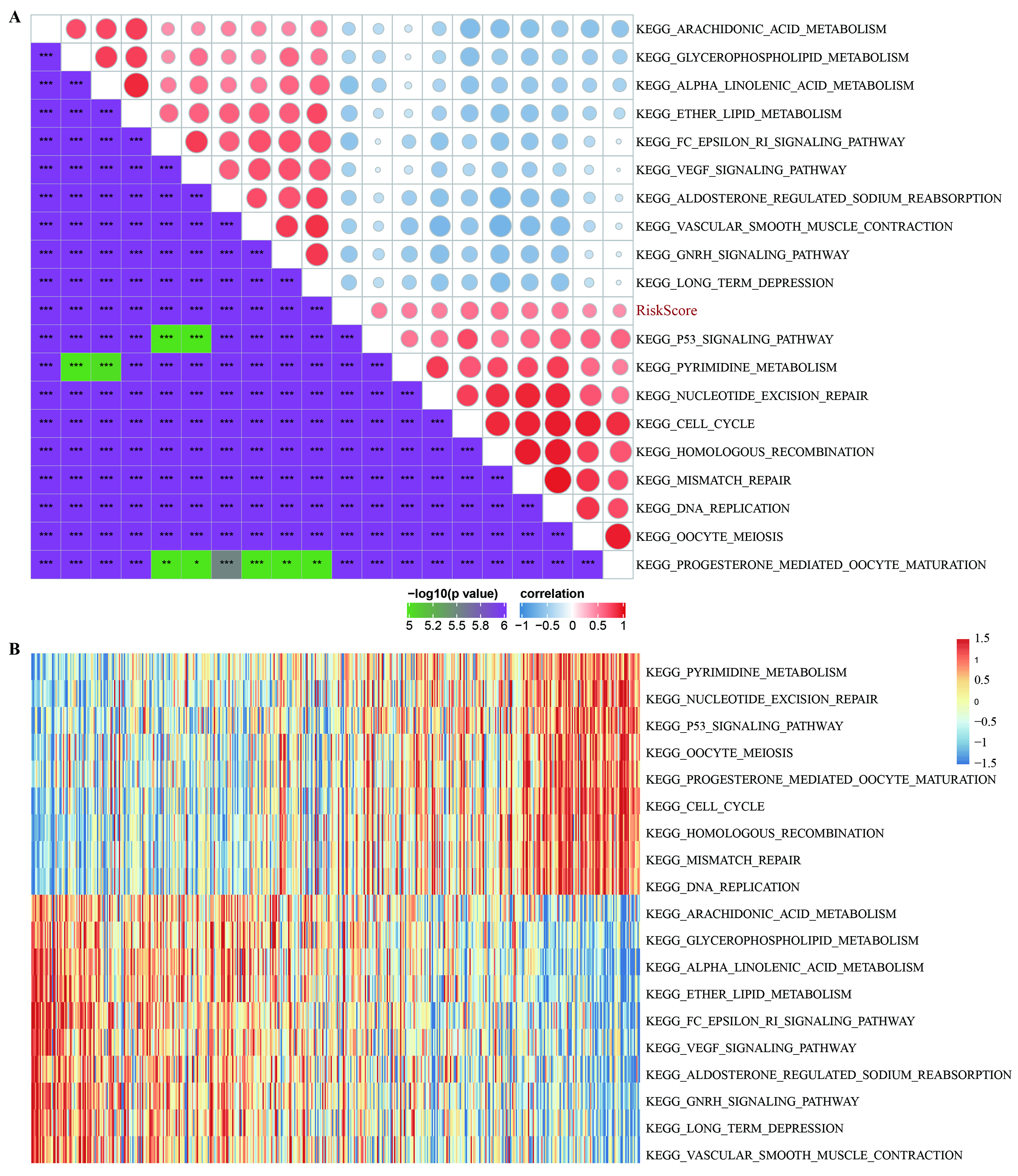
To observe the relationships between the risk scores and biological functions of different samples, GSEA was used to calculate the scores of different functions for each sample as well as correlations between these functions and risk scores. Correlation scores > 0.4 were considered to show positive correlations. Nine pathways were positively correlated with risk scores, and 10 pathways were negatively correlated with risk scores (Supplementary Fig. 4A). The 19 most relevant KEGG pathways were selected for cluster analysis (Supplementary Fig. 4B) based on their enrichment scores. Tumor-related pathways, such as KEGG_P53_SIGNALING_PATHWAY, KEGG_CELL_CYCLE, KEGG_MISMATCH_REPAIR, and KEGG_DNA_REPLICATION, were activated as risk scores increased, while others, such as KEGG_ARACHIDONIC_ACID_METABOLISM, KEGG_GLYCEROPHOSPHOLIPID_METABOLISM, and KEGG_ETHER_LIPID_METABOLISM, were deactivated as risk scores increased.
Construction of nomogram
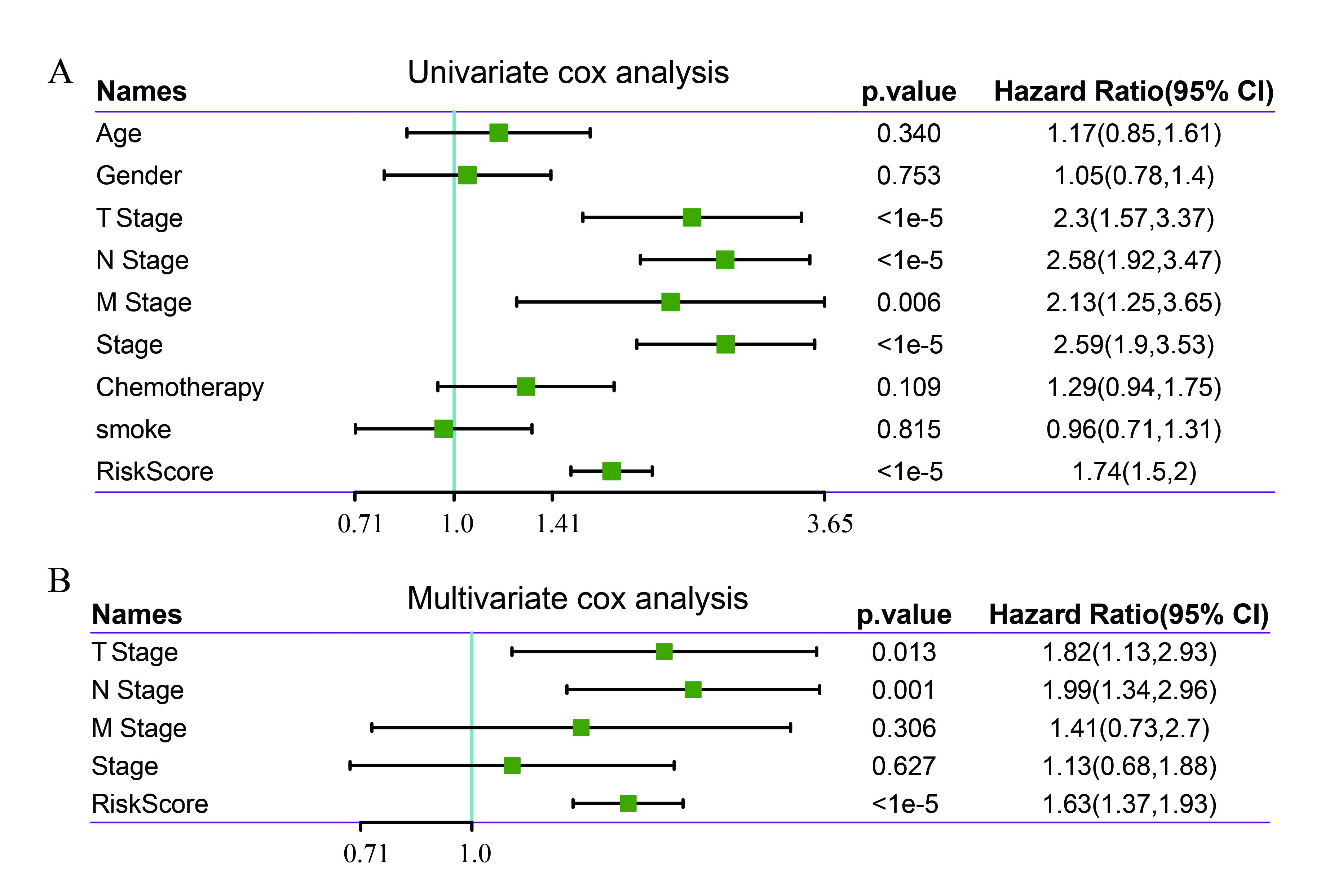
Univariate and multivariate Cox regression analyses were used to analyze the independence of the 5-gene signature model in terms of clinical applications. Univariate analysis results showed that TNM stage, stage, and risk scores were significantly correlated with survival time; multivariate Cox regression analysis results showed that risk scores (HR = 1.63, 95% CI = 1.34–2.96, p < 1e-5) and N stage (HR = 1.99, 95% CI = 1.34–2.96, p < 0.001) were independent prognostic risk factors (Supplementary Fig. 5A, B). A nomogram was constructed based on the significant variables of multiple factors (Fig. 6A), and the results showed that risk scores had the greatest effect on survival prediction, suggesting that the 5-gene signature was a good predictor of survival. Furthermore, by using calibration curves to evaluate the accuracy of the model (Fig. 6B), it was observed that the calibration curves at 1, 3, and 5 years were close to the standard curve, suggesting that the model had good prediction performance. Moreover, decision curve analysis was used to evaluate the model’s reliability (Fig. 6C), and the results showed that the benefits of risk scores and the nomogram were significantly higher than those of the extreme curve, and the effect of the nomogram was higher than the effects of T stage, N stage, and risk scores, which were close to the extreme curve, suggesting that risk scores and the nomogram had good clinical applicability.
Comparison of risk model with other models
To prove the superiority of our model, 3 risk models, including an 8-gene signature (Li) (Li et al.,, 2018), a 3-gene signature (Yue) (Yue et al.,, 2019), and a 3-gene signature (Liu) (Liu et al.,, 2018), were chosen to compare with our 5-gene signature. To make the models comparable, we calculated the risk score of each LUAD sample in the TCGA dataset by the same method, and we evaluated the ROC curve of each model. Z-score transformation of risk scores was performed. The samples with risk scores > 0 after Z-score transformation were divided into the high-risk group, and samples with risk scores < 0 after Z-score transformation were divided into the low-risk group. The survival curves were plotted. The results showed that all 3 models could significantly classify the high- and low-risk groups into prognostic categories (Fig. 6E, G, I). However, the AUCs of the ROC curves of the 3 models were lower than those of the 5-gene signature at 1, 3, and 5 years in the TCGA dataset (Fig. 6D, F, H). These results showed that our model had good clinical predictive power.
Expression of 5 genes in 33 pan-cancers
The box diagram showed that MS4A1 was significantly highly expressed in LUAD, HNSC, and kidney renal clear cell carcinoma, while in bladder carcinoma, colon adenocarcinoma, KICH, and READ tumors, MS4A1 was significantly lowly expressed (Fig. 7A). Compared with normal samples, KRT6A and MELTF showed significantly high expression in most cancer types, including LUAD (Fig. 7B, C), while CRTAC1 was expressed lowly in most cancer types, including LUAD (Fig. 7D, IRX5 was significantly highly expressed in breast cancer, CHOL, colon adenocarcinoma, kidney renal papillary cell carcinoma, liver hepatocellular carcinoma, and READ tumors, while in KICH, kidney renal clear cell carcinoma, lung squamous cell carcinoma, and PRAD, IRX5 was significantly lowly expressed (Fig. 7E).
Analysis of prognosis of genes in pan-cancer
We found that the MS4A1 gene is a protective gene in more than half of the tumors, which means, patients with high expression of MS4A1 have a better prognosis. KRT6A and MELTF genes are risk genes in most tumors including LUAD. CRTAC1 and IRX5 genes are significant protective genes in LUAD, while CRTAC1 gene is a high-risk gene in LUSC (Fig. 8A-E).
Clinical validation and gene set enrichment analysis of 5 genes
The results showed that in the Oncomine database, CRAT1 was lowly expressed in 12 LUAD studies, MS4A1 and MELTF were highly expressed in 1 LUAD study, KRT6A was highly expressed in 2 LUAD studies, and IRX5 showed no significant expression in any LUAD study (Fig. 9A-E). In The Human Protein Atlas database, the immunochemistry results of the 5 genes were analyzed, but only 4 genes (MS4A1, KRT6A, MELTF, CRTAC1) had protein expression data. The results showed that the expression levels of MS4A1, KRT6A, and MELTF in tumor tissues were higher than in normal tissues, while CRTAC1 expression in tumor tissues was lower than in normal tissues (Fig. 9F-I). At the same time, we found that the expression levels of 5 genes were significantly expressed in pan-cancer immune subtypes (Fig. 9J). Gene set enrichment analysis showed that MS4A1, KRT6A, and CRAT1 genes were both enriched in the HALLMARK_IL2_STAT5_SIGNALING pathway, and IRX5 and MELTF gene were both enriched in the HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION pathway. In addition, the KRT6A gene and CRAT1 gene were also enriched in the HALLMARK_KRAS_HALLMARK_UPRAS pathway (Fig. 9K-O).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}