Study design
This trial is a prospective, single-center, two-arms, double blinded, non-inferiority randomized controlled trial. The study is initiated by Peking University First Hospital, and its design has been completed in strict accordance with the SPIRIT 2013 statement (see Additional file 1 for the SPIRIT Checklist).
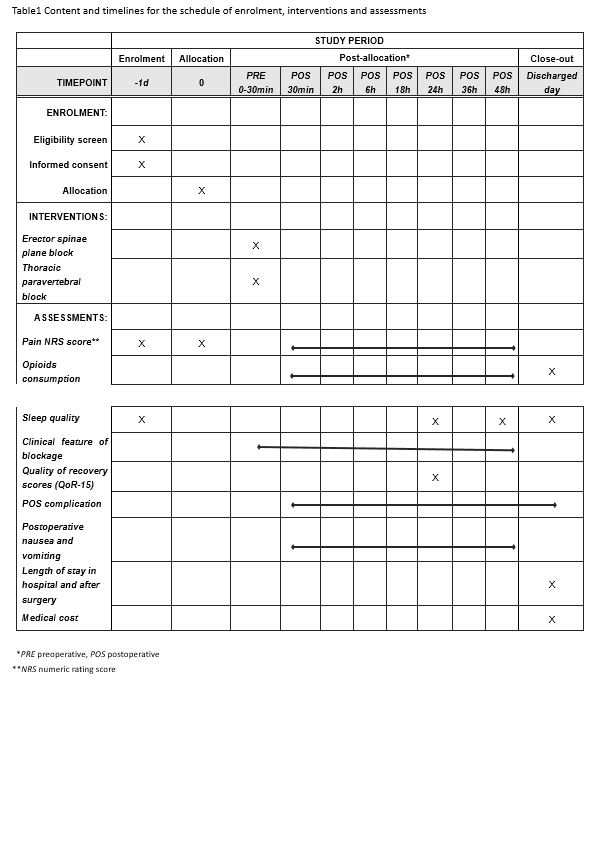
The protocol of this trial has been approved by the Clinical Research Ethics Committee of Peking University First Hospital (registration number: 2019-333) on 29 March 2020 with the latest version 1.1. This trial has been registered on Chinese Clinical Trials registry (identifier: ChiCTR2000031916). We will strictly adhere to the Good Clinical Practice guidelines and the Declaration of Helsinki during the whole period of the study. Flowchart diagram of the study is illustrated in Fig. 1, and the SPIRIT figure of enrolment, interventions, and assessments is presented in Table 1.
Participants
On the day before surgery, patients will be screened by X L according to inclusion/exclusion criteria. If the operation is on Monday, the screening will be performed on the previous Friday. Written informed consent will be obtained from each participant before the enrolment. Patients will be instructed how to self-assess pain severity via the Numeric Rating Scale (NRS, an 11 points scale where 0=no pain and 10=most severe pain).
Inclusion criteria
Participants meet all of the following criteria will be enrolled in this study:
(1) Between 18 and 75 years of age;
(2) Plan to undergo laparoscopic nephroureterectomy;
(3) Agree to receive regional nerve block and postoperative intravenous controlled analgesia (PCIA);
(4) American Society of Anesthesiologist (ASA) physical status classification I-III.
Exclusion criteria
Participants will be excluded if they have any of the following conditions:
(1) Refuse to participate in this study;
(2) BMI >35kg/m2 or <15kg/m2;
(3) Combined with severe comorbidities, including but not limited to renal dysfunction (Creatinine >442μmol/L or requirement of renal replacement therapy), liver dysfunction (Child-Pugh Class C) and heart failure, or ASA physical status classification ≥IV;
(4) Contraindication of deep nerve block, including but not limiting to be allergic to anesthetic drug, coagulation disorder, infection at the injection site;
(5) Chronic opioids dependence or chronic pain over 3 months;
(6) Unable to communicate preoperatively due to severe dementia, language barrier or neuropsychiatric disorder;
(7) Unable to perform nerve block procedure due to difficult anatomy found by ultrasound scan.
Randomization, allocation concealment and blinding
Randomization
The SAS 9.3 software package (SAS Institute, Cary, NC, USA) is used to generate random numbers with a block size of 4 by biostatisticians who will not participant in the statistical analysis of the data. The random sequence will be sealed in consecutively numbered opaque envelopes, and kept by the study coordinator. Participants will be randomly divided into two groups according to the 1:1 ratio of ESPB group and TPVB group. A research coordinator (Z Z) is designated to distribute and preserve randomization result. The coordinator will open the envelopes for allocation according to the order of enrolment and prepare the study drug.
Blinding
The participants will be blinded to the allocation. Because the needle injection of ESPB and TPVB are very close, participants themselves could hardly detect the clinical differences. Anesthesiologists who perform the nerve block, take charge of intraoperative management are independent individuals. Only the random sequence number rather than the specific nerve block type will be recorded in the electronic anesthesia information management system (AIMS). Researchers (ZZ X, ZY L, X L) who do not take part in the nerve block and intraoperative management are designated to postoperative follow-up. Therefore, all research staffs are blinded to the allocation. Besides, trained anesthesiologist (LL S, H Z) who do not perform the block will be designated to evaluate the clinical features of the block objectively. When all case report forms (CRFs) are written and checked, the data will be monitored by the Clinical Research Ethics Committee of Peking University First Hospital. After the database is locked, allocation will be handed over and revealed to statistical analysis.
Intervention
Participants will be admitted to the preoperative preparation area 30 min earlier before transferring to operating room (OR). After establishment of standard monitoring and intravenous access, premedication with 0.02mg/kg midazolam and 0.08 μg/kg sufentanil will be administrated if necessary. Before regional nerve block, all patients will be positioned lateral decubitus with the operative side up and received ultrasound scan by two experienced anesthesiologists (D H, H K) who will not take part in perioperative management and follow-up. After being confirmed as suitable participants for nerve block, patients will receive ultrasound guided ESPB or TPVB block according to the randomized grouping allocation. The drug used for both interventions is the same with a bolus of 0.375% ropivacaine in the volume of 0.4ml/kg. Once the block is completed, the dermatomes of sensory loss will be assessed by ice-tactus tests every 5 minutes. Both the onset time and time to peak the block will be recorded. Decreased or loss off thermic sensation of any region will be regarded as successful sensory loss (The illustration for predefined regional distribution was attached in Additional File 2).
ESPB group
Patients in ESPB group will be scanned by the linear high-frequency probe firstly placed in sagittal orientation at the middle scapula line. Once the imaging of the 12th rib emerges, T12 spinous process (SP) will be traced by sliding the probe medially and then marked. The probe will be then traced cranially to locate the T10-T11 vertebral. After same marked method, the probe will be continually moved 3-5 cm laterally and rotated in transverse orientation to identify the muscle layers of erector spinae and transverse processes near the marked site.
After re-confirming the important anatomic structure including lumbar artery under ultrasound scan, a 22-gauge nerve block needle (80 mm, Stimuplex D, B.Braun, Germany) will be inserted in-plane through a medial-lateral direction. Once the needle tip arrives beneath the erector spinae muscle, 3ml of normal saline will be injected firstly to ensure correct positioning of the needle after aspiration. The prepared study drug will be then injected into this plane with aspiration every 5 ml per injection in case of accidental puncture of vessel or pleura. Successful study drug injection is defined as the appearance of a hypoechoic ellipsoid with well-defined margin beneath erector spinae muscle on ultrasonic view.
TPVB group
T10 and T11 vertebral will be located and marked as the same way as that mentioned above in ESPB group. The probe will then be moved 3-5 cm laterally to identify the paravertebral space as the target injection site. After probe being rotated into transverse orientation, a 22-gauge nerve block needle (80 mm, Stimuplex D, B.Braun, Germany) will be inserted in-plane through a medial-lateral direction. Once the needle threads the internal intercostal membrane and arrives in the paravertebral space, 3ml of normal saline will be injected firstly. If displacement sign of the pleura occurs, the prepared study drug will be then injected into the confirmed paravertebral space. Successful study drug injection is defined as the appearance of pleura displacement sign and hypoechoic ellipsoid in paravertebral space under ultrasonic view.
Anesthesia management and postoperative analgesia
Intraoperative anesthesia management is performed by an attending anesthesiologist and an assistant, none of whom has knowledge of the allocation. Prednisone methylprednisolone (40mg) is administered before anesthesia induction, followed by intravenous anesthesia induction with sufentanil (0.1-0.3 µg/kg), propofol (1-2mg/kg) and rocuronium (0.6mg/kg). Anesthesia is maintained by intravenous anesthesia (propofol target controlled infusion, TCI) combined with N2O inhalation to maintain sedation level within the range of 40-60 under bispectral index (BIS) monitor. Opioid analgesics (remifentanil TCI together with sufentanil intermittent administration on demand) are used to maintain analgesia. During the surgery, the fluid volume and infusion speed are adjusted according to hemodynamic monitoring conditions to maintain the blood pressure within 20% of the baseline values. All surgical procedures are performed by fixed surgical team members, and the pneumoperitoneum pressure is often maintained at 12-16 mmHg as usual. After the surgery, standardized patient controlled intravenous analgesia pump is applied for each patient, which was established with 1.25 µg/ml sufentanil and programmed to deliver 4-ml boluses with a lock-out interval of 10 minutes and no background infusion rate. Patients who are enrolled for this trial will be trained to use a patient controlled intravenous analgesia pump before surgery.
Endpoints
Primary endpoint
The primary outcome in this trial is a joint endpoint of cumulative 24h opioid consumption after surgery and average pain NRS score at 24th h (valued the average from NRS at rest and with movement at 24th h) postoperatively.
Secondary endpoint
(1) cumulative opioid consumption at other different time (0.5h, 2h, 6h, 18h, 36h and 48h postoperatively) after surgery, as well as the PCIA pump the time interval to the first bolus demand, the numbers of requirement and administered boluses in PCIA pump.
(2) somatic and visceral pain NRS scores both at rest and with movement at preset timepoints (0.5th h, 2nd h, 6th h, 18th h, 36th h and 48th h) postoperatively.
(3) besides PCIA pump, demand of other rescue analgesic (observed at preset different timepoint, including the time interval to the first rescued analgesic in PACU or in ward).
The other endpoints are defined as follows:
(1) clinical features of blockage including time-consumption for the intervention, the onset time and time to peak of the block, dermatomal distribution of sensory loss, predefined regional distribution of sensory loss, times of puncture, and the adverse events.
(2) quality of recovery scores (QoR-15) will be assessed by a QoR-15 questionnaire [22] face-to-face at 24h after surgery.
(3) sleep quality scores on the nights of the first 2 days after surgery (assessed with NRS scores, 0=best sleep quality whereas 10=worst sleep quality).
(4) the incidence of postoperative vomiting and nausea score assessed with NRS scores (0=no nausea whereas 10=severe nausea) at the following timepoints (0.5th h, 2nd h, 6th h, 18th h, 24th h, 36th h and 48th h) postoperatively.
(5) the incidence of postoperative moderate to severe pain.
(4) overall incidence of postoperative complication (observation period: 48h after surgery, classified by Clavien-Dindo Classification) [23].
(5) time to first ambulation and flatus (observation from the end of surgery).
(6) length of postoperative hospital stay and total length of hospital stay.
(7) anesthesia cost and total hospitalization cost.
Safety consideration
We will strictly adhere steps to minimize the risk of adverse events which might be caused by regional nerve block. First, we will scan eligible patients before the enrolment, and exclude those who are difficult for TPVB and ESPB from anatomical point of view. Both blocks will be performed under directly ultrasound guiding by experienced anesthesiologists. Second, the local anesthetic we used for regional block is 1.5mg/kg ropivacaine (0.4 ml/kg with 0.375% in our study), which has already been proved to be safety in previous studies [24, 25]. Third, regional nerve block will be performed only after intravenous access and standard monitoring are established, therefore we can detect any adverse events timely. Lastly, participants will be continuously monitored till they are transferred to the general ward after surgery. Researchers will follow up the participants at preset timepoints after surgery, and all adverse events occurred during this period will be carefully managed and severe adverse events will be reported to the Clinical Research Ethics Committee as soon as possible. If the patient's harm level meets the insurance claims, payment will be arranged as soon as possible.
Data collection and management
Baseline data will be collected, including age, gender, birth date, education years, weight and height. Co-morbidities as well as important laboratory tests and instrumental examination will be documented. Preoperative recent sleep quality will be assessed with Pittsburgh Sleep Quality Index (PSQI) [26]. Intraoperative data, including duration of anesthesia and surgery, surgical information, anesthesia medication including opioid consumption and fluid balance will be documented. Pneumoperitoneum pressure will be documented as an average throughout the operation. Outcome data will be evaluated and recorded according to the follow-up plan to ensure all timepoints data are noted.
Based on the original observation records of the participants, the data are collected into the case report forms timely, completely and correctly. All data will be kept confidentially. Data entry will be performed in a double-input and double-check way with the REDCap (Research Electronic Data Capture) database system developed by Vanderbilt University. The data will be monitored and sampled regularly in the trial by the Clinical Research Ethics Committee, and the database will be locked after the electronic data is checked. After data entry and verification as required are completed, the case report forms will be filed in numbered order and kept in a specific filing cabinet. We have no plan in interim analysis until the target sample size is achieved.
STATISTICAL ANALYSIS
Sample size calculation
The primary endpoint of this non-inferiority trial is the cumulative 24h opioid consumption after surgery and average pain NRS score at 24th h postoperatively.
In a pilot investigation of our patients, the mean (± standard deviation) of the cumulative 24h opioid (sufentanil) consumption after laparoscopic nephroureterectomy was about 25.7 (±2.0) μg with TPVB, and 35 (±14.0) μg with non-block respectively. The non-inferiority margin was set as 5 (unit: μg; clinical practice treated it as an acceptable difference) [27]. With a significance level of α=0.05 and a power of 1-β= 90%, the sample size required to detect difference was 77 patients in each group.
A 1-1.3 points difference in pain NRS scores is usually considered as acceptable subjective pain discrimination [28]. When the standard deviation of the pain NRS score was assumed as 2.5 and the non-inferiority margin of NRS was 1 score [29], 70 samples were estimated per group.
Hence, the greater sample calculated above was 77 participants per group. Considering a dropout rate of 5% and technique failure rate of 3%, we finally planned to enroll 83 participants per group (a total of 166 participants). Sample size was estimated by PASS software (version 11.0; NCSS PASS, Utah, USA).
Endpoints analysis
Baseline characteristic will be compared according the data distribution and type. In addition to graph method for qualitative analysis, Shapiro-Wilk test is also used for quantitative analysis in review of data distribution. Continuous data with normal or approximate normal distribution is expressed as mean ± standard deviation, while continuous data with non-normal distribution is expressed as median (interquartile, IQR). Continuous data will be tested by independent sample t test or Mann-Whitney U test according to data distribution. Categorical data is expressed as numbers (percentages) and tested by chi-square test /Fisher's exact test. Time-to-event data is analyzed by the Kaplan-Meier estimator, with the difference between groups tested by the log-rank method. Two-tailed p values of less than 0.05 is regarded as statistically significant. All statistical analyses are performed with the SPSS 25.0 statistical package (IBM SPSS Inc., Chicago, IL, USA).
Primary endpoint
Non-inferiority
Cumulative 24h opioid consumption and average pain NRS score at 24th h after surgery are the primary joint endpoints, which will be analyzed by “joint hypothesis test”. In this analysis, the intervention group could be considered effective as long as neither of the two endpoints is inferior. The confidence interval method will be used to perform the non-inferiority test and the result will be expressed as effect size (difference) (95%CI). If the effect size upper limit of the one-side 95%CI is smaller than 5 in cumulative 24h opioid consumption, we will conclude that ESPB is non-inferiority to TPVB; in addition, when it is smaller than 1 in average pain NRS scores at 24th h after surgery, ESPB group will be also considered non-inferiority.
Superiority
If noninferiority is conducted for both primary outcomes, the superiority of the corresponding comparison will be evaluated for each outcome using an overall α of 0.025 with Holm-Bonferroni correction for testing both outcomes (upper limit of 97.5%CI smaller than predefined margin for most significant outcome and upper limit of 95%CI for the other). If superiority is detected on at least either cumulative 24h opioid consumption or pain NRS scores, the ESPB group will be claimed to be better than TPVB group.
Secondary endpoint
For the opioid consumption and pain NRS scores at other different timepoint after surgery, repeated measure ANOVA (analysis of variance) will be performed if normality and homogeneity of variance and sphericity hypothesis (Mauchly’s test) are met. If not, one-way ANOVA and its correction (Greenhouse-Geisser coefficient correction and Huynh-Feldt coefficient correction) as well as generalized estimated equation (GEE) model will be performed. As for inter-group comparison, independent sample t test will be used for comparison if the distribution accords normality assumption; if distribution is skewed as well as not independent at different timepoint, the difference will be determined by Mann-Whitney U test.
Outcome analyses will be performed in the intention-to-treat (ITT) population. A per-protocol (PP) analysis was also performed for the primary endpoint. Statistical analysis will be performed by professional biostatisticians. All conclusion will be based on the original data.
{kind=link}