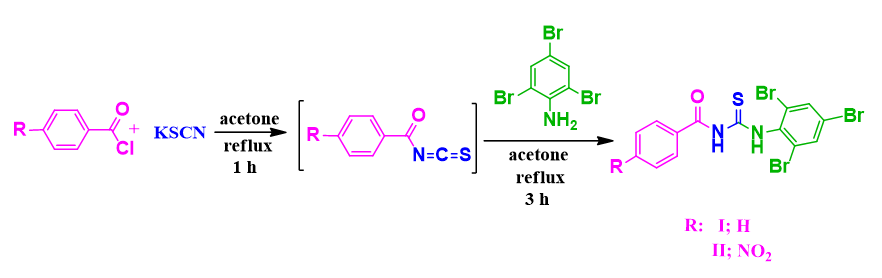
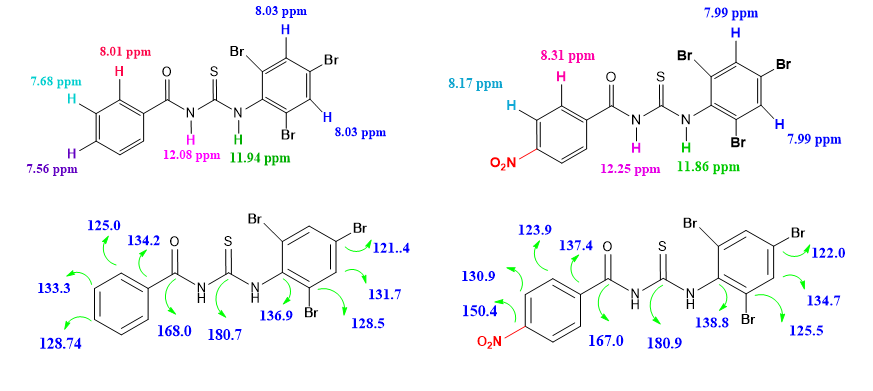
Two new compounds N-Benzoyl-N'-tribromophenyl thiourea (C14H9Br3N2OS) (I) and 4-Nitrobenzoyl-N'-tribromophenyl thiourea (C14H8Br3N3O3S) (II) were synthesized and characterized by 1HNMR, 13CNMR, IR, and structural (XRD) techniques. The molecular geometry, vibrational frequencies of the title compound (I, II) in the ground state have been calculated by using the density functional theory (DFT) method with B3LYP / 6–311G(d,p) basis set and compared with the experimental data. The calculated results show that the optimized geometries can well reproduce the crystal structural parameters. A detailed vibrational spectral analysis has been carried out and assignments of observed fundamental bands have been proposed on the basis of peak positions. The scaled theoretical frequencies show very good agreement with experimental values. Frontier molecular orbitals energies (HOMO&LUMO), energy gap (ΔE), global chemical reactivity parameters such as ionization potential (IP), electron affinity (ΔE), chemical hardness (η) and chemical softness (σ), etc. have been calculated, the sites of electrophilic and nucleophilic regions where the molecular interactions likely to happen are identified. Besides, molecular electrostatic potential (MEP) and thermodynamic properties of the title compounds were investigated by theoretical calculations.
{kind=link}
{kind=link}