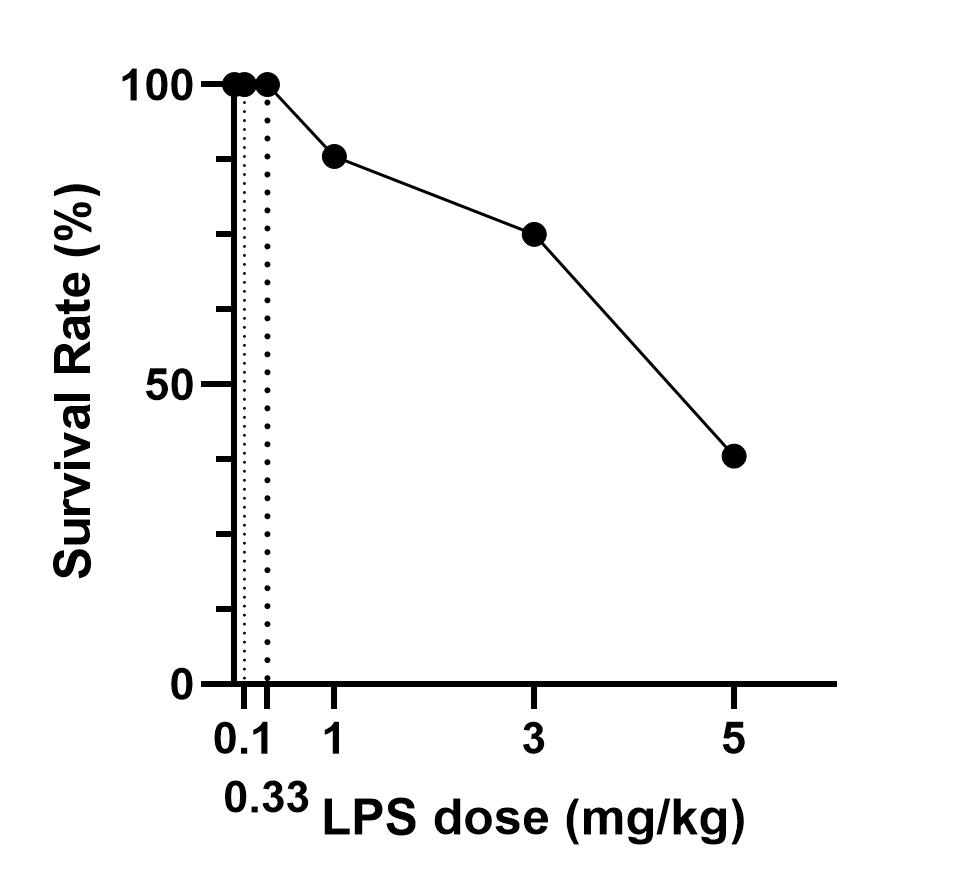
LPS-induced M1 polarization has been broadly validated in rodents, but doses of LPS vary between studies[10,12-14,25]. Yang injected mice i.p. with 1 mg/kg LPS for 4 hours to induce M1 polarization, and observed an upregulation of CD16 and CD86, which marks M1, and downregulation of M2 markers such as Arg1, Ym1, and TGF-β1 in the brain[10]. Fenn[13] and Liao’s[14] data showed that a 0.33 mg/kg LPS injection could increase the expression of M1 markers such as IL-1β, IL-6, TNF-α, and iNOS. Liu et al. treated mice with an even lower LPS dose (0.1 mg/kg) for 24 hours to induce M1 activation, and they detected upregulation of TNF-α, IL-1β, IL-6, CD86, CD16, and CD32 in the brain[12]. In our experiments, we showed that systemic LPS with doses ranging from 0.1 to 5 mg/kg induced M1 activation in both the brain and spinal cord. As the LPS dose increased, the number of activated M1 markers (≥ 2-fold) increased slowly, with 13 of 22 activated at 5 mg/kg LPS. In particular, M2 markers presented a similar increasing trend and degree as M1. Thus, we infer that systemic LPS lasting 24 hours with a dosage ranging from 0.1 to 5 mg/kg may not be a proper choice to induce M1 polarization. On the one hand, it did not make microglia polarize into M1 more than M2; on the other hand, only half of the M1 genes were activated, with less than 25% upregulated at the most reported dose of 0.1 mg/kg. In a study comparing 4 hours and 24 hours of LPS treatment, a weaker activation of M1 markers CD86, iNOS, and IL-1β was detected at 24 hours compared to the 4-hour group[13]. Whether a shortened stimulation of LPS contributes more to M1 polarization than M2 polarization is not clear. Moreover, in Wang and Zhao’s study[19,26], small LPS doses (0.75 and 0.25 mg/kg, respectively) were administered i.p. for 7 days to induce cognitive deficits by activating M1 microglia.
LPS has been recently reported to induce A1 activation. Studies by Liddelow[11], Zamanian[7], and Kano[27] all used systemic LPS 5 mg/kg to induce A1 polarization, and Zhang[28] selected 0.83 mg/kg LPS to activate A1 astrocytes to create murine depression-like behavior. Our results showed that even a small intraperitoneal LPS dose (0.1 mg/kg) could activate more than half of the A1 markers. As LPS increased, more than 80% of A1 markers were activated in both the brain and spinal cord. Furthermore, fewer A2 markers were activated, especially in the brain, indicating that LPS contributes to superior A1 polarization, thus supporting it as a good inducer to activate A1 astrocytes.
Besides the upregulated M1 and A1 genes induced by LPS, we also found that M2 and A2 markers were significantly increased, as demonstrated by our co-immunofluorescence of elevated S100a10 with GFAP certificates. The dichotomy of M1 and M2, or A1 and A2, is an oversimplified conceptual framework. M2, which is often induced by IL-4 can be further characterized into several main states[29]: M2a with alternate activation and involvement in repair and regeneration; M2b with an immunoregulatory phenotype; and M2c with an acquired-deactivating phenotype. Chhor et al. found that the majority of M1 markers (10/13) and M2b markers (8/13) were maximally expressed after 12 hours of exposure to LPS. Moreover, M2c genes (IL-10, Il-4Rα, and SOCS3) were also increased in enriched microglia 4 hours after LPS injection[30]. Similarly, we found that more than half of M2 genes, especially M2a genes, were activated when the dose of LPS injection was larger than 0.33 mg/kg, and most reached a maximum at LPS 5 mg/kg. Identically, although less sensitive to LPS compared with A1, A2 was also activated, particularly in the spinal cord. A2 is often induced by MCAO and upregulates many beneficial inflammatory factors, such as hypoxia-induced factors, IL-6 and IL-10[11,31]. Markers of A2 could also be activated by LPS, although they were more sensitive to MCAO. For example, as A1 markers, H2-D1 was induced 30-fold by LPS, but only 3-fold by MCAO, and Serping1 was induced 6.5-fold after MCAO and 34-fold after LPS[11]. Our results showed that Serping1 was induced 34-fold in the cortex, 111-fold in the hippocampus, and 55-fold in the spinal cord under LPS 5 mg/kg, indicating a difference in tissues induced by LPS. Overall, LPS exposure could induce not only a pro-inflammatory response, but also anti-inflammatory action. However, the question remains whether the transient M2 or A2 polarization is a self-limiting reaction that is beneficial for regeneration or just a recovery failure.
Genes that are induced by both neuroinflammation and ischemia were classified as pan-reactive (A-pan) genes[15], different from the specific genes (A1) induced by neuroinflammation. Our research showed that, with increasing LPS dose, A-pan markers presented a similar trend to A1, especially in the cerebral cortex and spinal cord. It should be noted that the dichotomy of A1, A2, and A-pan is an oversimplified conceptual framework, and the status of astrocytes may include a battery of different, but overlapping, functional phenotypes. Although LPS is often used as a classical reagent to induce A1 astrocytes, it could activate other astrocyte phenotypes like A-pan, which may show an overlapping function with A1. We infer that selective blocking of A1 formation could be discounted by the function-similar A-pan, and this requires further study.
In addition to the various LPS doses, gene changes in different tissues were also detected. The cortex and hippocampus were preferred when studying LPS-induced impairments in spatial learning and memory. In Wang et al.’s study, mice were administered i.p. with LPS (250 μg/kg) once daily for 7 days, a 2-fold increase in TNF-α was found in the cortex and hippocampus by ELISA, and increases were nearly 3-fold for IL-1β in the cortex and 2.5-fold in the hippocampus[19]. Consistent with this, our results showed that when exposed to 0.33 mg/kg LPS, TNF-α mRNA was increased 2-fold both in the cortex and hippocampus, while IL-1β was raised by 19-fold in the hippocampus and 36-fold in the cortex, which may correlate with the increased LPS dose-induced pro-inflammatory state. In particular, more markers of microglia and fewer astrocytes were activated in the hippocampus than in the cortex and spinal cord. These data remind us that activated genes induced by LPS vary not only with LPS dose, but also tissue type and stimulation time. The complex signals in the lesion microenvironment determine the polarization of glial cells.
Our results showed that several markers, such as Marco, Ym1, and C3, presented a significant dose-response relationship with LPS increase in any tissue. TLR4 is an important receptor of LPS, and the interaction of TLR4 with adaptor MyD88 leads to the activation of downstream NF-κB and subsequent production of pro-inflammatory cytokines[32]. With increasing LPS, more pro-inflammatory cytokines were released. Whether these dose-dependent markers are involved in inflammation production or could reflect the degree of inflammation or glial cell polarization remains to be elucidated.
Lectin Ym1, a well-established marker of murine M2, is a secretory protein strongly induced by IL-4 and IL-13. The protective action of Ym1 expression after stroke has been demonstrated in several studies. Fumagalli et al. found increased Ym1 levels with reduced infarct sizes 24 h after transient MCAO in CX3CR1-deficient mice[33]. Barbera-Cremades showed the expression of Ym1 was associated with the inhibition of macrophage proliferation and is induced both by anti-inflammatory signal adenosine and ATP, and pro-inflammatory signal LPS[34]. This is consistent with Lee’s study, which showed that LPS significantly increased Ym1 in the anterior cortex, hippocampus, and entorhinal cortex[35]. This raises questions regarding how the upregulation of Ym1 and other M2 or A2 markers induced by LPS is regulated by pro-inflammatory stimuli, and why mRNA levels of Ym1 are increased in AD patients and animal models of amyloid deposition, which are typically considered to be associated with a pro-inflammatory (M1 or A1) cytokine environment. These results also suggest a more complex set of microglia and astrocyte phenotypes than the dipolar M1/M2 or A1/A2 characterization, and indicate that LPS-induced neuroinflammation may be mediated by distinct activation subtypes. Meanwhile, the classical markers of M1, such as CD86 and M2 marker CD206, increased with LPS in the cortex and hippocampus, but not in the spinal cord, suggesting that the particular markers of phenotypes should vary with tissues.
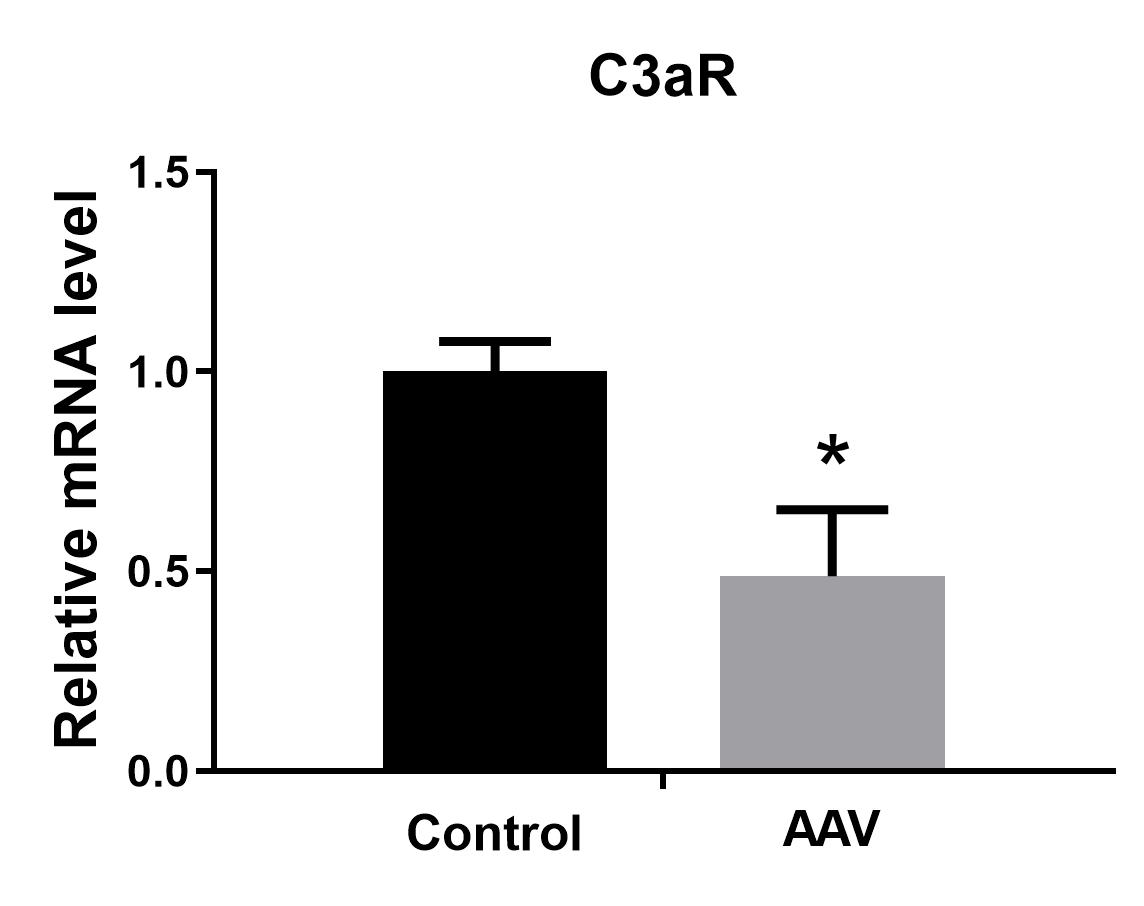
Downregulation of C3aR in astrocytes affected the status of activated markers A1 and A2 induced by LPS. Only Ggta1 (4-fold) and Srgn (2-fold) of A1 were upregulated, while more markers of A2, including S100a10, Clcf1, Emp1, Sphk1, Slc10a6, and especially Ptx3 (10-fold) and CD14 (7.4-fold), were elevated, indicating that A2 polarization dominated the reactive astrocytes when C3aR in astrocytes was knocked down. Interestingly, C3aR downregulation in astrocytes under LPS stimulation also affected microglial activation, such as decreasing IL-1β, Stat1, and TNF-α in M1 and increasing CD206, IL-1ra, and Clec7a in M2. The significant upregulation of IFN-γ and downregulation of TNF-α reflect a dominant anti-inflammatory state, consistent with the superior activation of M2 and A2. We infer that the superior M2 activation may be caused by the A2 polarization. During development, astrocytes can sense subtle changes in neurons to induce the production of C1q in neuronal synapses, which interacts with the microglial C3aR to prune the neuronal synapses through the classic cascade complement pathway[23]. In the context of Alzheimer’s disease pathology, overproduction of C3 from astrocytes can simultaneously communicate with microglial C3aR and neuronal C3aR to dynamically regulate microglial phagocytosis and impair dendritic morphology, as well as synaptic function, subsequently resulting in deterioration of cognitive function[22]. Meanwhile, reactive A2 astrocytes could also stimulate more active M2 microglia to produce an anti-inflammatory microenvironment.
Limitations
There were several limitations to our study. Our focus was on the dose relationship between LPS and gliocyte phenotypes, so we mainly used RT-PCR to quantify this. Morphology data on gliocyte polarization were partly shown, which could provide more information about polarization. We employed three variables—dose, phenotypes, and tissues—in our study, and did not include time variants. Considering that astrocytes are often activated following microglia, polarization changes with time need to be detected in future studies. Though we did not show the classical one-way ANOVA or Student’s t test statistical results which we have done, the presented figures could help better interpretate the multiple-dimensional RT-PCR results.
In conclusion, our results showed that systemic LPS activates not only M1 and A1, but also M2, A2, and A-pan. Only a few markers of the 72 genes showed a significant dose response to LPS, which may correlate with the degree of inflammation caused by LPS. No more A1 astrocytes were polarized at 5 mg/kg LPS than at 3 mg/kg LPS in the brain and spinal cord. In addition, the downregulation of C3aR in astrocytes contributed to more A2 and M2 polarization in the spinal cord when exposed to LPS.
{kind=link}
{kind=link}