Optimization of the spatial organization in multi-functional heterogeneous biocatalysts
The enzyme cascade is composed of two NAD+-dependent alcohol dehydrogenases from Bacillus stearothermophilus (ADH1) and horse liver (ADH2) to synergistically oxidize 1,5-PD to δ-valerolactone (lactone) via its corresponding lactol intermediate (tetrahydro-2H-pyran-2-ol). As mentioned above, the pool of NAD+ is replenished by an oxygen-dependent NADH oxidase from Thermus thermophilus HB27 (NOX) coupled to a catalase from bovine liver (CAT) that depletes the hydrogen peroxide generated as a by-product of NOX. Finally, a lactonase from Sulfolobus islandicus (LAC) hydrolyzes δ-valerolactone to 5-HP (Fig. 1a). In a previous work, this multi-enzyme system showed very promising conversion yields by optimizing the enzyme ratio to overcome the bottleneck of the reaction (the oxidation of the lactol into the lactone).18 However, we encountered issues when trying to scale up the reaction (from 2 mL to 10 and 25 mL) due to enzyme instability. To overcome these issues, we (co)-immobilized the 5 free-enzymes involved in the cascade on the tri-functional support described above (AG-Co2+/A/G) following different spatial configurations (Table 1). First, we individually immobilized all enzymes on the same support resulting in five monofunctional heterogeneous biocatalysts (HB1 to HB5). In this configuration each enzyme is immobilized on a bead different from the others, naming this spatial distribution as D1 (Entry 1, Table 1). Secondly, ADH1 and ADH2 were immobilized separately on AG-Co2+/A/G but co-immobilized with NOX and CAT resulting in biocatalysts HB6 and HB7, respectively, and finally mixed with LAC immobilized on AG-Co2+/A/G by its own (HB5) to assemble the configuration D2 (Entry 2, Table 1). Thirdly, NOX and CAT were co-immobilized with both ADHs (ADH1 and ADH2) on AG-Co2+/A/G yielding the heterogeneous biocatalyst HB8 that was mixed with HB5 containing only LAC to assemble configuration 3, D3 (Entry 3, Table 1). Finally, the five enzymes were sequentially co-immobilized on AG-Co2+/A/G to prepare biocatalyst HB9 with a configuration 4, D4 (Entry 4, Table 1). All HBs were incubated with 1 M glycine upon 2 h of enzyme immobilization to block the remaining aldehydes which do not intervene in the enzyme attachment.
Table 1
Immobilization parameters of enzymes bound to AG-Co2+/A/G with different spatial distribution, enzyme loads, and polymer coatings
|
Entry
|
Distribution
|
Heterogeneous biocatalyst
|
Enzyme
|
Enzyme load
(mg·g− 1)
|
Ψ a
(%)
|
Recovered activity b
(U·g− 1) /(%)
|
|
1
|
D1
|
HB1
|
ADH2
|
15c
|
100
|
0.42 (23)
|
|
HB2
|
NOX
|
0.41
|
81
|
0.67 (5)
|
|
HB3
|
CAT
|
0.010c
|
40
|
107 (25)
|
|
HB4
|
ADH1
|
5
|
100
|
1.52 (11)
|
|
HB5
|
LAC
|
1.49
|
98
|
0.35 (21)
|
|
2
|
D2
|
HB6
|
ADH2
|
7.4 c
|
99
|
0.5 (26)
|
| |
NOX
|
0.21
|
33
|
0.21 (15)
|
| |
CAT
|
0.004
|
56
|
109.7 (26)
|
|
HB7
|
ADH1
|
2.45
|
98
|
1.19 (28)
|
| |
NOX
|
0.12
|
94
|
0.67 (5)
|
| |
CAT
|
0.004
|
65
|
127.3 (30)
|
|
HB5
|
LAC
|
1.49
|
98
|
0.35 (21)
|
|
3
|
D3
|
HB8
|
ADH2
|
4.20c
|
99
|
na (na)
|
| |
NOX
|
0.13
|
57
|
0.40 (20)
|
| |
CAT
|
0.004
|
38
|
237 (61)
|
| |
ADH1
|
1.26
|
100
|
0.66 (23)
|
|
HB5
|
LAC
|
1.49
|
98
|
0.35 (21)
|
|
4
|
D4
|
HB9
|
ADH2
|
3.0c
|
100
|
na (na)
|
|
NOX
|
0.12
|
65
|
0.53 (30)
|
|
CAT
|
0.0023
|
19
|
343 (36)
|
|
ADH1
|
1.0
|
100
|
0.9 (50)
|
|
LAC
|
0.30
|
85
|
0.33 (46)
|
|
5
|
dD5
|
HB10
|
ADH2
|
3.0c
|
100
|
na (na)
|
| |
|
NOX
|
0.14
|
79
|
0.50 (26)
|
| |
|
CAT
|
0.12c
|
99
|
104 (8)
|
| |
|
ADH1
|
0.99
|
99
|
1.08 (50)
|
| |
|
LAC
|
0.32
|
100
|
0.18 (23)
|
|
6
|
dD5
|
HB11
|
ADH2
|
3.0c
|
100
|
na (na)
|
| |
|
NOX
|
0.15
|
82
|
0.51 (26)
|
| |
|
CAT
|
0.12c
|
99
|
120 (9)
|
| |
|
ADH1
|
1.0
|
100
|
1.04 (47)
|
| |
|
LAC
|
0.32
|
100
|
0.18 (23)
|
|
7
|
dD5
|
HB12
|
ADH2
|
3.0c
|
100
|
na (na)
|
| |
|
NOX
|
0.66
|
74
|
1.25 (17)
|
| |
|
CAT
|
0.87c
|
73
|
770 (35)
|
| |
|
ADH1
|
1.0
|
100
|
1.07 (49)
|
| |
|
LAC
|
0.32
|
100
|
0.20 (25)
|
|
8
|
dD5
|
HB13
|
ADH2
|
3.0c
|
100
|
na (na)
|
| |
|
NOX
|
0.72
|
80
|
1.7 (20)
|
| |
|
CAT
|
1.14
|
95
|
116 (5)
|
| |
|
ADH1
|
1.0
|
100
|
1.5 (53)
|
| |
|
LAC
|
0.32
|
100
|
0.26 (32)
|
|
a Immobilization yield, Ψ = (immobilized activity/offered activity) × 100. b (%) Recovered activity of the immobilized enzyme is defined as the coefficient between the specific activity of the immobilized enzymes and the specific activity of the soluble ones. c Total protein content in a semi-purified enzyme extract. dD5 is the distribution where the 5 enzymes are co-immobilized and co-localized at the outer region of the same bead.
|
These four spatial distributions imply that intermediates must follow different interparticle diffusion pathways toward the final product. Accordingly, in D1 all intermediates must travel from one particle to the other to be processed by their corresponding enzyme (Fig. 1b). In contrast, as D2 segregates each oxidation step but confines the NAD+ recycling and H2O2 removal, the only intermediates forced to travel between particles are the lactol and the lactone (Figure S1a). In the case of D3, only the lactone must diffuse between different particles to be hydrolyzed by LAC (Figure S1b). Finally, as D4 confines the five enzymes inside the same particle, interparticle transport of intermediates is not needed to complete the cascade target product (Fig. 1c). Expectedly, the immobilization parameters for each enzyme varied depending on whether the enzymes were individually immobilized or co-immobilized together (Table 1). This phenomenon was already reported for ADH1, whose recovered activity when immobilized alone is different from the activity recovered when co-immobilized with other enzymes.21
Once the 9 different HBs (HB1-9) were prepared, they were mixed to assemble the multi-enzyme systems with the corresponding spatial distribution (D1-D4) in reaction, keeping a protein mass ratio of 1:3:0.18:0.012:0.32 for ADH1:ADH2:NOX:CAT:LAC, respectively. Monitoring the reaction courses, we observed that D1 and D4 converted 75% of 1,5-PD yielding up to 60% of 5-HP in 24 h (Figs. 1b and 1c), whereas D2 and D3 only reached a 35% 5-HP yield after the same time (Figures S1a and S1b). Remarkably, Fig. 1d shows that all enzymes co-immobilized on the same particle (Entry 4, Table 1) transform 1,5-PD into 5-HP 1.6 times faster than all enzymes physically segregated into different particles (Entry 1, Table 1). Since the oxidation of the lactol intermediate is the rate-limiting step in this cascade due to the high apparent KM of ADH2 towards it,22 its greater accumulation using the HB9 with D4 configuration may speed-up the lactone production, thus contributing to improve the overall throughput of the cascade when using this spatial configuration. As D1 and D4 configurations present the most promising results in terms of product yield and productivity, we discarded D2 and D3 configurations for further studies.
Operational stability of the heterogeneous multi-functional biocatalyst with different spatial configurations.
Due to the promising performance of HBs under D1 and D4 spatial configurations, we tested their operational stability in consecutive batch reaction cycles by assessing the cascade coupling (Fig. 2). This latter parameter is defined as the produced mol of 5-HP per consumed mol of 1,5-PD, where the ideal system would reach a coupling efficiency value of 1, indicating a perfect cascade orchestration where the substrate (1,5-PD) and intermediates (lactol and lactone) are quantitatively converted into the final target product (5-HP). Although HB9 with configuration D4 (Entry 4, Table 1) outperforms HB1-5 in configuration D1 (Entry 1, Table 1) for the first cycle, the cascade coupling efficiency was higher with D1 than with D4 in the second and third consecutive cycles, pointing out that the 5 enzymes co-immobilized together are less stable than those separately immobilized on the same support (Fig. 2a). To understand the lower operational stability of the co-immobilized system (D4), we investigated the catalytic efficiency of each cascade step using a set of spectrophotometric assays that allowed us to determine the activity of the diol oxidation (Figure S2a), the NADH oxidation (Figure S2b), the hydrogen peroxide accumulation (Figure S3) and the ω-hydroxy acid production (Figure S4). Figure 2b shows that HB9 in configuration D4 is 3 and 4 times faster for the diol oxidation and NADH recycling, respectively, than the configuration D1 using HB1-5. These results match the reaction time courses (Fig. 1), supporting the higher overall throughput of this cascade when it is catalyzed by the 5-enzymes co-immobilized on the same porous particle of AG-Co2+/A/G. However, the D4 spatial configuration accumulates H2O2 5 times faster than the configuration D1, suggesting that CAT in the co-immobilized system cannot match the activity of the NOX. As H2O2 is a liaison for enzymes, its accumulation in the reaction catalyzed by HB9 in configuration D4 explains the system inactivation during the process. Despite the unsatisfactory operational stability, the high efficiency of the co-immobilized biocatalysts encouraged us to enhance its operational stability by optimizing its capacity to remove the H2O2 formed in situ, without limiting the NADH recycling.
Optimization of the intraparticle spatial distribution and loading of 5-enzymes co-immobilized system to maximize its performance.
To investigate why hydrogen peroxide is accumulated when the cascade is catalyzed by HB9 in configuration D4, we studied the intraparticle spatial distribution of a co-immobilized enzyme system by confocal laser scanning microscopy (CLSM) using enzymes labeled with compatible fluorophores for colocalization studies. Figures 3a and S6 show that four of the co-immobilized enzymes are located at the outer surface of the particle, whereas NOX is located at the inner regions of the beads. This spatial distribution agrees with the spatial distribution found for the five enzymes individually immobilized on this support (Figure S5), as previously reported.20 The intra-particle segregation of NOX and CAT may explain their impaired activities. Thus, the hydrogen peroxide can be accumulated during biotransformation as it is produced at large distances (inner regions of the particle) from where it can be removed (outer regions of the particle), damaging the co-immobilized enzymes.
To improve the hydrogen peroxide removal, we optimized the spatial distribution of NOX by tuning its immobilization kinetics, colocalizing all 5 enzymes at the outer surface of the beads. As previously demonstrated, high enzyme immobilization rates yield immobilized enzymes located at the outer parts of microbeads,23 while slowly immobilized enzymes are uniformly distributed across the beads. The immobilization rate can be easily controlled by either adding immobilization competitors or modifying the immobilization buffer and/or conditions. To favor a more rapid NOX immobilization rate and enable its localization at the outer surface of the beads, we performed its immobilization on AG-Co2+/A/G in the presence of a gradient of NaCl concentration (0, 0.1, 0.5, and 1 M). 1 M NaCl was needed to locate NOX at the outer surface of the support, colonizing the outer 2% radius of the beads (2.5 µm on average) (Figs. 3b and S7). Figures 3c and S8 show the CLSM images that demonstrate the colocalization of the 5 enzymes at the outer region of the same bead. When 1 M NaCl is added to the immobilization buffer, the Manders´s coefficients of NOX regarding the other enzymes (Fig. 3d, Table S1) determined from the CLSM images confirm that NOX colocalizes with the rest of the enzymes to a higher extent than when NaCl was not added. As the support is positively charged, it can repel NOX, slowing its immobilization. Hence, we hypothesize that the chlorides will act as counter ions to the positive amine groups of AG-Co2+/A/G, minimizing the repulsion and consequently immobilizing NOX faster on the outermost surface. The outer localization of NOX brings it closer to CAT, enhancing their cooperative action, but also increasing the NAD+ recycling efficiency as the oxygen transport from the bulk to NOX is facilitated. The HB bearing the 5 enzymes colocalized at the outer surface of the beads will be now referred to as HB10 with a distribution 5 (D5) (Entry 5, Table 1). Furthermore, we corroborated that this new spatial location of NOX negligibly affects the immobilization pattern of the other enzyme members of the cascade.
Next, we evaluated the effect of the intraparticle NOX spatial distribution on biocatalyst productivity (Fig. 4a) and operational stability (Fig. 4b). First, we observe that the localization of NOX at the outer surface of the beads increases 5-HP titer upon 24 h reaction and maintains the chromatographic product yield (CY ≈ 70%) constant for 3 consecutive batch cycles unlike the HB9 (Entry 4, Table 1) where NOX is localized in the deeper surface of the porous support (Fig. 4b). Unfortunately, the HB10 suffered operational inactivation upon the 4th operational cycle, observing a CY decay of 20%. To further increase the operational stability of HB10, we incubated the immobilized enzymes for longer times (16 h at 4 ºC) before the blocking step to fabricate HB11 (Entry 6, Table 1). Longer immobilization times pursue promoting the formation of more attachments between the residues at the enzyme surface and the aldehydes of AG-Co2+/A/G, to ultimately improve the enzyme stability as reported elsewhere.13 Nevertheless, the increase in the immobilization time neither enhances the efficiency nor the operational stability of the HB11. Finally, to further optimize the performance of HB10 biocatalyst, we increased the load of NOX and CAT by 4.7 and 7.25 times, respectively, resulting in a heterogeneous biocatalyst named HB12 with distribution D5 (Entry 7, Table 1). The specific activity of the immobilized NOX in HB12 decreased 1.8 times due to the higher protein density within the porous beads. Previous studies support the fact that NOX is less catalytically efficient at high protein loads, suggesting that protein crowding negatively affects the performance of this enzyme.8 Despite this activity reduction, HB12 converted 100% 1,5-PD to yield 80% 5-HP after 24 h. Surprisingly, we observed 20% production of the 5-oxopentanoic acid (5-OPA), indicating the overoxidation of the target 5-HP. This product overoxidation hints at a very efficient NAD+-recycling system that boosts the oxidative activity of both coimmobilized dehydrogenases (ADH1 and ADH2). Regarding the operational stability, the excess of immobilized CAT drove to a less operationally stable biocatalyst as the product yield dramatically decayed to 10% upon re-using this biocatalyst in 5 consecutive batch cycles. In summary, the overall efficiency and operational stability of the 5-enzyme heterogeneous biocatalyst are optimized by localizing NOX at the outer surface of the beads and increasing the NOX and CAT loading in the biocatalyst, yet longer immobilization times negligibly improve the biocatalyst performance. To note, higher enzyme loads resulted in an HB less operationally stable. To understand whether the operational inactivation of HB12 relies on the enzyme lixiviation due to an excessive load, we performed an SDS-PAGE analysis of HB10-12. This electrophoretic analysis reveals that enzyme lixiviation similarly occurred in all of them (Figure S9), thus operational inactivation may be triggered by the enzyme subunit leaching (quaternary structure disassembly) under reaction conditions, among other causes.
Improvement of the multi-functional heterogeneous biocatalysts by cationic polymer coating.
As most immobilized enzymes on HB12 are lixiviated during their operational use, we decided to stabilize their quaternary structure by polymer coating using polyallylamine (PAH). The enzyme coating with this cationic polymer enhances the performance of dehydrogenases and oxidases as previously reported by our group.24 To that aim, after sequentially co-immobilizing the 5 enzymes with the optimal spatial distribution and enzyme loading, we coated them with PAH, fabricating a new version of HB12 named HB13 (Entry 8, Table 1). The primary amines of PAH react with the remaining aldehyde groups of the support not involved in enzyme attachment, acting as an ionic macromolecular crosslinker of enzyme subunits and as a blocking agent to make the support inert after the immobilization processes. This polymer coating increased the recovered activity of ADH1, ADH2, and NOX, suggesting that the aminated polymer has a stabilizing effect on the quaternary structure of the immobilized enzymes. SDS-PAGE analysis (Figure S9, lanes 9 and 10) confirms that stabilizing effect since enzyme subunits are lixiviated to a lower extent when enzymes are coated with PAH.
Next, we tested HB13 for the stepwise oxidation/hydrolysis of 1,5-PD into 5-HP in one pot. As a result, HB13 achieves a CY of 100% using 10 mM substrate in only 3 hours in comparison with the 80% conversion achieved with the same biocatalyst but blocked with glycine (HB12) (Figs. 5a and 5b). When the substrate load was scaled up to 20 mM, HB13 reached 90% CY in 6 hours (Figure S10a). Then, we studied the operational stability of HB13 by submitting it to consecutive recycling in 24 h batch cycles. Figure 5c shows how the PAH-coated heterogeneous biocatalyst was operationally stable for 4 consecutive cycles, while the substrate conversion decayed below 50% when using the non-coated HB12. This tendency is reflected also in the 5-HP yield along the cycles (Figure S10b).
After successfully assembling a productive and stable multi-functional heterogeneous biocatalyst (HB13), we scaled the batch reaction volume up to 30 mL with 3.3% (w:v) biocatalyst load and monitored the product titer, the oxygen concentration, and the pH along the reaction course (Figure S11). As a pH decay occurs concomitantly to the 5-HP production, we manually kept the pH constant to a value of 8 by NaOH titration during the operation. The performance of HB13 in this scaled cascade is notorious as we achieved 80% 5-HP yield after 96 h operation, which means a product titer of 16.4 mM (c.a 0.5 mmol), a maximum volumetric productivity of 0.053 g·L− 1·h− 1 and a specific mass productivity of 1.1 mg·gHB13−1·h− 1. Furthermore, we observed a 6% reduction of the oxygen saturation in the reaction bulk during the first hour of the reaction. This oxygen depletion is related to the high rate of the first oxidative step (1,5-PD to lactol) associated with a very efficient NAD+ regeneration system that concomitantly consumes molecular oxygen by the action of NOX. Then, the oxygen level increases until it reaches its steady saturation concentration (22%). This experimental evidence supports a very efficient coupling between ADH1 and its cofactor regeneration system (NOX/CAT).
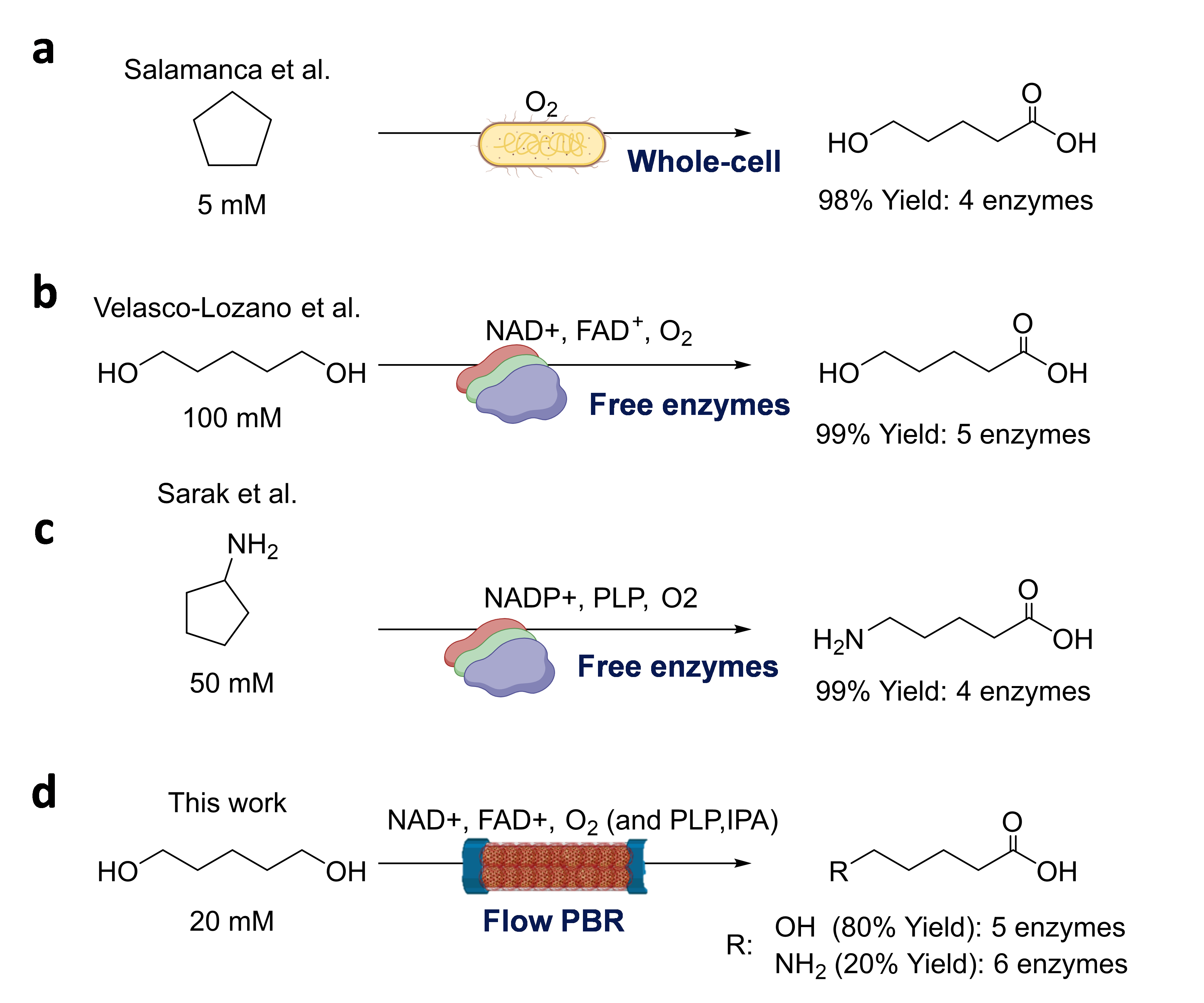
Implementation of the optimal multi-functional heterogeneous biocatalyst in packed bed reactors for continuous synthesis of 5-HP.
In our efforts to intensify the process, we decided to integrate HB13 into a packed bed reactor (PBR). This PBR packed with 1 g of HB13 was first flushed with 10 mM 1,5-PD at 0.02 mL·min− 1 showing no product formation. The UV-Vis spectra of samples collected from the PBR outlet demonstrated that the pool of redox cofactor was NADH (Figure S12), indicating the premature cascade halt due to an inefficient NAD+ recycling. Interestingly, the outlet samples were colorless indicating that FAD+ was either absorbed to the surface of HB13 as reported for other heterogeneous biocatalysts coated with cationic polymers, or reduced to FADH2 but not reoxidized due to the absence of oxygen.25 This latter hypothesis is supported by the poor solubility of oxygen in the aqueous media (0.25 mM) and the lack of aeration within the PBR, explaining why the PBR fails to transform 1,5-PD into 5-HP due to the inefficient FAD and NAD+ in-situ regeneration. To overcome such a limitation, we flushed the PBR with an air-saturated solution but unfortunately neither did we detect the product.
Inspired by previous works from Nidetzky’s26 and Turner’s27 groups who managed to release soluble oxygen in a flow reactor flushing hydrogen peroxide in the presence of catalase, we decided to follow a similar approach to enhance NAD+ recycling driven by NOX (Fig. 6a). As HB13 integrates CAT, we run the PBR with this multi-functional heterogeneous biocatalyst using 20 mM 1,5-PD and varying H2O2 concentration at 0.02 mL·min− 1 (Fig. 6b). At 45 mM of H2O2, we achieved a maximum substrate conversion and product yield (CY) of 80% and 60%, respectively, determined by GC. However, at 90 mM H2O2, we observed a dramatic decay in the CY likely due to the harmful effect of the hydrogen peroxide on enzyme stability. As expected, we also observed a linear correlation between the pH drop and the product titer at the reactor outlet, due to the accumulation of higher concentrations of the target ω-hydroxy acid (Figure S13).
Once the optimal H2O2 concentration was found, we next challenged the PBR to a flow rate ramp to find its productivity limits. Figure 6c shows that the maximum product CY is achieved at the lowest flow rate, giving as a result the lowest STY. In contrast, the highest STY productivity occurred at a flow rate of 0.1 mL·min− 1 at the expense of product titer with a CY as low as 18%. Thus, we selected 0.01 mL·min− 1 and 45 mM H2O2 as the optimal conditions to operate the PBR. Under these conditions, we achieved 8.3 mM 5-HP in 150 min (residence time) with a STY = 0.76 ± 0.07 g·L− 1·h− 1 and a specific mass productivity of 0.76 mg·gHB13−1·h− 1 (Fig. 6c). This latter parameter is slightly lower than that achieved with the 30 mL batch reactor under the same reaction conditions, suggesting that oxygen supply is still more efficient in a stirred tank than in an H2O2-fed PBR. Moreover, the 1H-NMR of the sample collected directly from the outlet of the PBR shows a purer product than the sample separated from the batch process catalyzed by the free enzyme system (Figure S14). Under the optimal reaction conditions described above, the PBR was continuously operated for 44 h (Fig. 6d). The STY was maintained during the first 12 h of operation and afterward steadily decreased to zero after 44 h of operation, detecting no product in the reactor outlet. Post-used and ex-situ activity assays revealed that the ADH activity of 44 h operated HB13 was 2 times lower than its fresh counterpart. In contrast, NOX retained 90% of its initial activity in the exhausted biocatalysts. Therefore, the STY decay during the continuous operation is linked to the inactivation of the co-immobilized ADH1 and ADH2.
Finally, we calculated the turnover frequency (TOF) of each reaction step by analyzing the profiles of products and intermediates at the PBR outlet. Figure 6e shows that the rate-limiting step is the intermediate oxidation of the lactol to the lactone as the TOF of ADH2 is 7- and 16-fold lower than those of ADH1 and LAC, respectively. Remarkably, the generation of oxygen in situ is the most catalytically efficient step assuring the efficient recycling of the redox cofactor.
Implementation of two telescoped PBRs for the biotransformation of 1,5-PD into 5-amino pentanoic (5-AP) acid.
To further exploit the potential of this multi-functional heterogeneous biocatalyst, we studied its application in the biosynthesis of 5-aminopentanoic (5-AP) acid. This ω-amino acid has gained attention for its potential use in nylon synthesis.28,29 To achieve this, we combined HB13 with a previously reported multi-functional heterogeneous biocatalyst (HB14)29 for the conversion of 5-hydroxypentanoic (5-HP) acid into 5-AP (Fig. 7a). We assembled HB14 by co-immobilizing ADH2, NOX, and a transaminase from Halomonas elongata (HewT) on methacrylate beads. This support was functionalized with epoxy and aldehyde groups for the irreversible immobilization of ADH2, NOX, and HewT and further coated with polyethyleneimine (PEI) to improve the biocatalyst stabilization (Figure S15a). In contrast to our previous work,29 we selected here an H2O2-forming NADH oxidase (NOX) encouraged by its excellent behavior as part of HB13 in flow reactors fed with H2O2. Immobilization yields were higher than 75% for all co-immobilized enzymes, whereas the recovered activities ranged 3.5–65% depending of the enzyme, being NOX the one that recovered the lowest activity (Figure S15b).
Once HB14 was fabricated, we packed HB13 and HB14 in two different PBRs and telescoped them for the continuous biotransformation of 1,5-PD into 5-AP (Fig. 7a). Then, the continuous flow reaction was carried out in circulation mode at 0.02 mL·min− 1, allowing the unreacted substrates to contact both PBRs for longer times. Remarkably, no additional air supply was needed for the PBR2 as the segmented air-liquid flow was naturally generated from the PBR1 by the action of CAT (Fig. 7a, inlet). Isopropyl amine (IPA) was added to the reaction mixture as an amine donor for the last transamination reaction. Under these conditions, 95% of 1,5-PD was consumed and 3.5 mM of 5-AP was produced in 21 h (Fig. 7b), which means 3 reactor cycles for PBR1 and PBR2, with a total residence time of 7.25 and 9 h, respectively. Longer reaction times (up to 72 h) failed to increase the 5-AP titer, mainly due to the inactivation of the immobilized ADHs, whose activity decayed more than 70% upon their continuous operation (Table S2). To enhance the efficiency of this 6-enzyme 2-reactor system for the complete conversion of 1,5-PD into 5-AP in flow, we anticipate ongoing efforts. These include replacing ADHs with more robust ones, refining the immobilization strategy, and increasing the excess of amine donors.
{kind=link}