The structure of the S.album mitochondrial genome
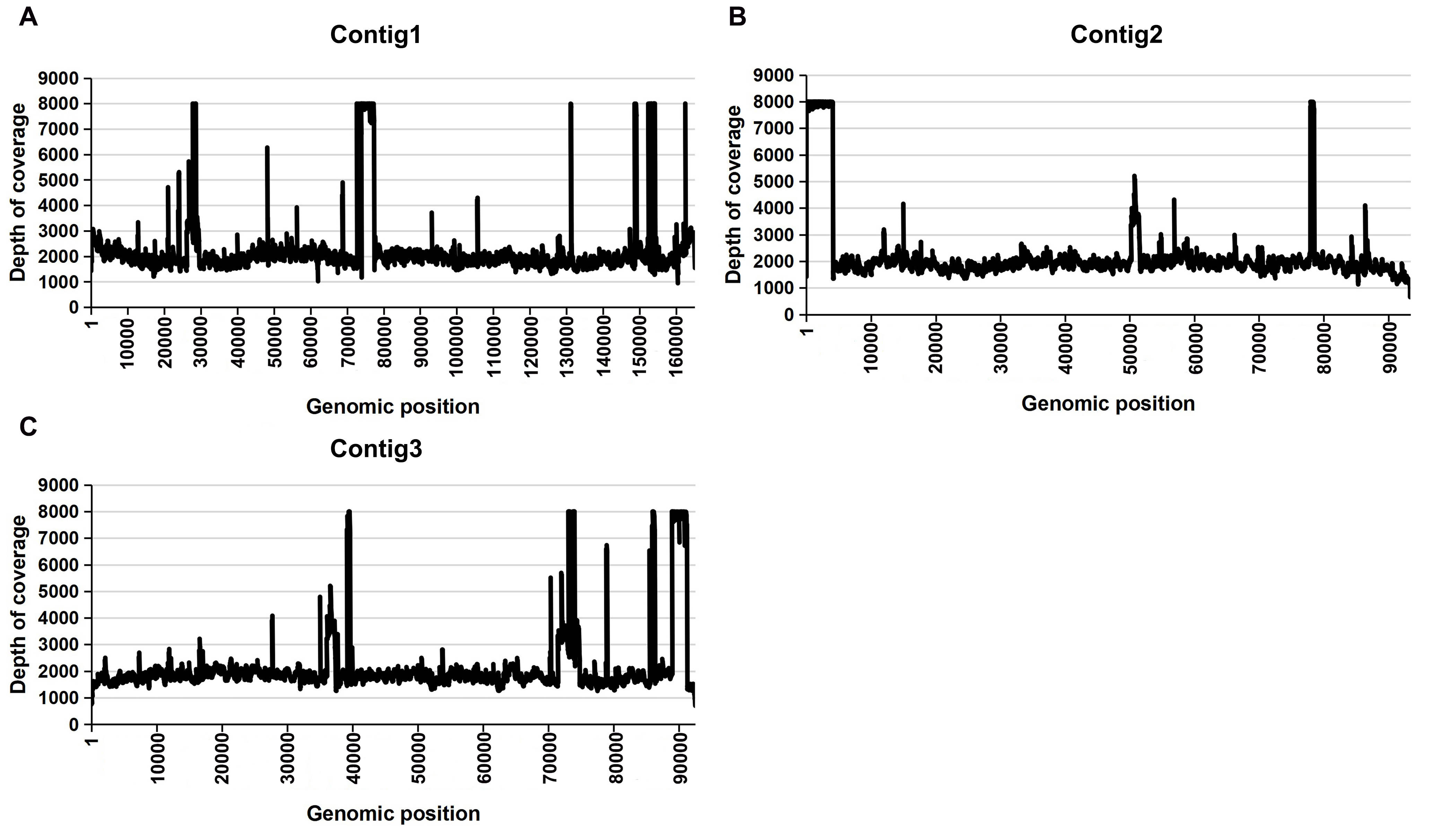
In our research, the genome structures resulting from both short-read and long-read assemblies show a high level of consistency. The Bandage software was utilized for visualizing the graphical assembly of the mitochondrial genome. As shown in Fig. 1A, there are 3 nodes, and in this visualization, each node represented a contig that had been assembled. These nodes had regions of overlap with each other along the connected lines. Among these three contigs, contig1 is 165,122 bp, contig2 is 93,430 bp and contig3 is 92,491 bp. The graphic results indicated that contig1 (c1) can form a circle with contig2 (c2) and contig3 (c3) respectively. We then aligned all the raw data to the sequences we had assembled (Figure S1). The comprehensive coverage of every base on the three contigs by short reads, as illustrated in Figure S1, confirms the accuracy of our assembly.
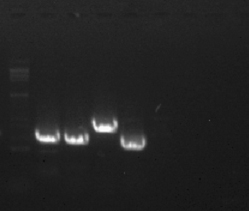
To verify the rightness of the conformation, we performed PCR experiments and Sanger sequencing. The position of the four sets of primers is illustrated in Fig. 1D. To validate the accuracy of paths p1, p2, p3, and p4, we employed the primer pairs (F1 + R1), (F2 + R2), (F3 + R3), and (F4 + R4) respectively, as shown in Fig. 1C. The PCR outcomes revealed bands with expected lengths (Fig. 1B), and the subsequent Sanger sequencing (Figure S2) affirmed the veracity of these paths. The untreated electropherogram is shown in Figure S3. In a word, the mitochondrial genome of S.album is a complex branched structure consisting of three contigs. And for convenience, we will analyze these contigs individually in the next paragraphs.
Mitochondrial genome annotation
We artificially annotated these three contigs as circular for convenience. There are 34 unique mitochondrial protein-coding genes (PCGs) in S.album mitochondrial genome, comprising 24 core genes that are unique and 10 unique variable genes. The gene information is detailed in Table 1. Within the core genes, we find ATP synthase genes like atp1, atp4, atp6, atp8, and atp9, as well as cytochrome c biogenesis genes such as ccmB, ccmC, ccmFC, and ccmFN; nine genes belonging to the NADH dehydrogenase category, specifically nad1, nad2, nad3, nad4, nad4L, nad5, nad6, nad7, and nad9; along with three genes associated with cytochrome c oxidase, namely cox1, cox2, and cox3.; and others including mttB, matR, and cob. On the variable genes side, the 10 unique genes consist of three large ribosomal protein subunits: rpl5, rpl10, rpl16, five small subunits such as rps3, rps4, rps7, rps10, rps12, and succinate dehydrogenases sdh3 and sdh4. The genome also incorporates 26 tRNA genes, where 21 stand unique. Out of these, 13 are native to mitochondria, with others like trnH-GUG_ copy2, trnD-GUC, trnM-CAU, trnI-CAU, trnN-GUU, trnP-UGG_ copy2, trnW-CCA, trnA-UGC, and trnI-GAU being derivatives from plastids. Some tRNA genes, however, remain unknown origin due to their lack of homology with recognized organelle tRNA genes. For an in-depth insight, Table S1 lists gene locations, while Fig. 2 visualizes the mitochondrial genome maps. Importantly, all the genes we've identified in S.album, 15 contain introns, as shown in Table 1. And the accession number of S.album mitochondrial genome is OQ868374.
Table 1
Gene composition in the mitochondrial genome of Santalum album.
|
Group of genes
|
Gene Name
|
|
ATP synthase
|
atp1, atp4, atp6, atp8, atp9
|
|
Cytochrome c biogenesis
|
ccmB, ccmC, ccmFC*, ccmFN
|
|
Cytochrome b
|
cob
|
|
Cytochrome C oxidase
|
cox1, cox2**, cox3
|
|
Maturation enzyme
|
matR
|
|
Membrane transport protein
|
mttB
|
|
NADH dehydrogenase
|
nad1****, nad2****, nad3, nad4***, nad4L, nad5****, nad6, nad7****, nad9
|
|
Ribosomal small subunit
|
rps10*, rps12, rps3*, rps4, rps7
|
|
Ribosomal large subunit
|
rpl10, rpl16, rpl5
|
|
Succinate dehydrogenase
|
sdh3, sdh4
|
|
Transfer RNA
|
trnE-UUC (×2)*, trnH-GUG(×2)*, trnT-UGU*, trnV-UAC*, trnC-GCA, trnD-GUC, trnF-GAA, trnG-GCC, trnK-UUU, trnM-CAU, trnfM-CAU (×3), trnI-CAU, trnN-GUU, trnP-UGG (×2), trnQ-UUG, trnS-GCU, trnS-UGA, trnW-CCA, trnY-GUA, trnA-UGC*, trnI-GAU*
|
|
Ribosomal RNA
|
rrn18, rrn26 (×2), rrn5
|
|
Note: (×2) indicates that the gene had two complete copies, * indicates the number of introns that the gene contains
|
Repetitive elements
Microsatellites, also referred to as SSRs (simple sequence repeats), are typically sequences of 6 bp found in eukaryotic genomes. In S.album mitochondrial genome, we found 89 SSRs (Table S2). And these SSRs are dominated by tetramers, accounting for 44.94% (41) of all. Subsequently, we observed monomeric (8), dimeric (17), trimeric (20), and pentameric (3) SSRs. A depiction of the SSRs' spread across individual contigs can be found in Fig. 3. And 11 tandem repeat elements were also detected (Table S3). In the three contigs, we identified 242 dispersed repeat pairs that were 30 bp or longer. This set comprises 141 forward repeats and 101 palindromic repeats. Notably, no reverse repeats were observed (refer to Table S4). The quantity of dispersed repeats markedly surpasses that of SSRs and tandem repeats, with the majority of these repeat elements being shorter than 500 bp. Only five dispersed repeats are longer than 500 bp, the 3,325 bp palindromic repeat element is the longest dispersed repeat. The repeated units of the dispersed repeats appear both within individual contigs and across different ones. Summing up the lengths, the dispersed repeats total 18,978 bp, which constitutes 5.41% of the entire S.album mitochondrial genome. At last, the Circos package was used to visualize the dispersed repeats of S.album mitochondrial genome (Fig. 4). It's important to highlight that such repeats could not only instigate genome reconfiguration but also impact its overall size.
The results of Characteristic of Mitochondrial Plastid DNA sequences (MTPTs)
In the mitochondrial genomes of angiosperms, there's a frequent occurrence of sequence migration from both plastomes and nuclear genomes. In our investigation, we detailed the annotation of S.album's chloroplast genome and contrasted it with its corresponding mitochondrial genome. Through the use of the BLASTn program, we pinpointed 20 MTPTs that serve as a link between the genomes of these two organelles. These MTPTs cover 22,353 bp, which constitutes 6.37% of the mitochondrial genome and 15.52% of the plastome. Among them, MTPT12 stands out as the lengthiest at 4,109 bp, whereas MTPT15 is the briefest at just 37 bp. Upon annotating these MTPTs, it became evident that they all housed plastidial genes. Table S5 illustrates that, there are nine complete migrated tRNAs, and they are low in nucleotide substitutions. In these nine complete migrated tRNAs, four tRNA genes, namely trnP-UGG, trnA-UGC, trnW-CCA and trnI-GAU have moved in conjunction with MTPT12 and MTPT16. trnI-CAU (MTPT6), trnI-GAU (MTPT12), trnA-UGC (MTPT12) and trnM-CAU (MTPT20) maintain certain flanking regions in the mitochondrial genome within the fragment. Conversely, the other five complete tRNA genes (trnD-UGC, trnN-GUU, trnH-GUG, trnW-CCA and trnP-UGG) occupied the length of the MTPT, that is, almost no flanking regions were retained after the migration. Conversely, the transferred PCGs have seen some sequence loss, with only fragments being identifiable, barring atpE. This implies a potential loss of function for these PCGs. Figure 5 provides a visual depiction of the MTPTs.
The Prediction of RNA Editing Sites
In this part, we Used Deepred-mt tool, and we pinpointed 628 RNA editing sites across 34 mitochondrial PCGs, as detailed in Table S6. They are all C to U edits. Figure 6A visually show the forecasted RNA editing sites for PCGs. Notably, nad4 and ccmB stood out with 50 and 40 RNA editing sites respectively, ranking them the highest among the PCGs. It is interesting that these two genes also have the most RNA editing sites in many plant mitochondrial genomes. Next are ccmC, ccmFN, mttB, nad1, nad2 and nad7, they showcased over 30 editing sites. On the other end of the spectrum, rps10 and rps7 possess the least number of C to U editing sites, with only two each. Notably, the start codons for genes such as cox1, cox2, nad1, nad4L, and nad5 emerge through RNA editing, transforming ACG to AUG. Additionally, RNA editing establishes the stop codons for rps10 and ccmFC, converting CGA to UGA.
The impacts of RNA editing on amino acid alterations are depicted in Fig. 6B. Out of the editing instances, 602 led to changes in amino acids, signifying that non-synonymous substitutions account for 95.86% of these edits. Many RNA editing events resulted in the substitution of Pro (proline) and Ser (serine) by Leu (leucine) during translation,happening 147 times and 131 times. And the above two account for 46.18% in total. In contrast, only 26 synonym substitutions exist. It suggested that these broad RNA editing events are important for protein translation.
The Results of Collinear Analysis
To investigate the rearrangement in the mitochondrial genomes, we identify homologous blocks of collinearity identified using the BLASTn program. Figure 7 illustrates ribbons connecting two neighboring mitochondrial genomes, with each ribbon symbolizing a collinear block of highly similar sequences. And the mitochondrial genomes showed poor collinearity. There are many regions without homology between these mitochondrial genomes. These findings suggest significant genomic reconfigurations between S.album and its related mitochondrial genomes, underscoring the S.album mitochondrial genome is not conserved in genomic structure.
Phylogenetic Analysis
31 mitochondrial genomes of angiosperm species were used to construct a phylogenetic tree. Paropyrum anemonoides and Aconitum kusnezoffii were used as the outgroups. Table S7 displays the list of species and their respective GenBank accessions employed in the phylogenetic tree. Phylogeny analysis yielded an Maximum Likelihood treeexhibiting high support for the principal basal branches (Fig. 8). It was consistent with the latest classification of the Angiosperm Phylogeny Group (APG IV system), S.album is most closely phylogenetically related to T.maclurei, the two species belong to the order Santalales. Nevertheless, some plants on the tree did not receive high support. This may be related to the phenomenon of horizontal transfer of plant mitochondrial genes. Therefore, the phylogenetic tree constructed based on mitochondrial DNA does not necessarily represent the true evolutionary relationship. Mitochondria, as a relatively independent genetic system, may have conflicts with nuclear or chloroplast genomes in evolution.
{kind=link}
{kind=link}
{kind=link}