Animals
21-day-old Sprague-Dawley rats were purchased from SiPeiFu (Beijing) Biotechnology Co. All animals were housed in environmentally controlled cages with a 12 h/12 h light/dark cycle. After acclimatization, the rats in this experiment were divided into two parts for treatment, one part for in vitro experiments to collect ovarian granulosa cells for in vitro culture, and the other part for in vivo experiments with 5 IU of eCG injection. 48 h later, plasma samples were collected into heparin-spiked tubes and centrifuged at low speed (3000 rpm for 20 min). SD rats were euthanized using an overdose of pentobarbital (Beijing BioDee Co., Ltd., China), and their ovaries were dissected. We collected the supernatant (plasma) after centrifugation and stored it at a low temperature (-20℃). The ovaries were weighed, and their cross-sectional diameters were measured (n = 20). One side of the ovaries of SD rats (n = 10) was fixed in 4% paraformaldehyde for histological and immunohistochemical analyses, whereas the other side of the ovaries (n = 10) was immediately frozen in liquid nitrogen and stored at -80℃ for gene expression analysis. All procedures followed the Animal Care and Use Policy of the Ethics Committee of Beijing Forestry University and were approved (EAWC_BJFU_2023026).
Primary granulosa cell culture in vitro
The freshly obtained 21-day-old rat ovaries were placed in PBS to remove the perivitelline adipose tissue and oviducts, etc., and then incubated in 6 mM EGTA (Sigma, LOT: SLBG8546V)-M199 for 5 min, and then transferred to 0.5 mol/L sucrose-M199 for 15 min, and then finally transferred to fresh M199 medium (Gibcoo, LOT: 2333381). All antral follicles were punctured with a 27-gauge needle under a microscope, releasing granulosa cells and oocytes. The granulosa cells were then blown and mixed, and oocytes and other tissue debris were filtered with a 40 µm cell sieve. The collected granulosa cell suspension was centrifuged at 1000 rpm for 5 min to remove the supernatant. The cells were then resuspended with 1 mL M199 added with 10% FBS, and then viable cells were counted using the Taipan blue assay. After that, the cell suspension was diluted with M199 medium according to the experimental requirements and cultured in the six-well plates (Corning, 3335) within a 37℃ incubator with 5% CO2. The next day, after the cells were attached to the wall, the original culture medium was discarded, and the cells were washed three times with M199 medium cultured in serum-free M199 medium overnight, then treated with AdipoRon according to the experimental design.
Histology
Ovarian tissues soaked in 4% paraformaldehyde for 24 h were removed, washed three times with PBS for five minutes at a time, dehydrated in an alcohol-xylene gradient, and embedded in paraffin. Paraffin blocks were sectioned at 5 µm thickness. The paraffin sheets were laid flat on warm water and picked up with a bonded slide so that the sheet with the tissue was attached to the surface of the slide. The deparaffinized tissue sections were stained with hematoxylin-eosin. The stained sections were subjected to gradient dehydration and sealed with coverslips glued with neutral resin. The overall histomorphology, follicular distribution, and cell type of the prepared tissue sections were observed under the microscope.
Immunohistochemistry
Deparaffinized ovarian sections were incubated in a citrate buffer solution and then blocked with 10% goat serum. Tissues were incubated with primary antibodies including AdipoR1 (sc-46748, Santa Cruz Biotechnology, Santa Cruz, CA, USA), AdipoR2 (sc-46751, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Adiponectin (ab181281, Abcam Biotechnology, Abcam, Cambs, UK), GLUT1 (21829-1-AP, Proteintech Biotechnology, Hubei, China), GLUT2 (20436-1-AP. Proteintech Biotechnology, Hubei, China), GLUT3 (20403-1-AP, Proteintech Biotechnology, Hubei, China), GLUT4 (sc-53566, Santa Cruz Biotechnology Santa Cruz, CA, USA). BrdU (sc-6326, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Tissues were further processed with goat anti-rabbit IgG/HRP kit (KGOS60, KeyGEN BioTECH Biotechnology, Jiangsu, China), stained with diaminobenzidine solution (30 mg DAB, 150 mL 0.05 M pH 7.6 Tris HCl solution, 25 µL H2O2) and counterstained with hematoxylin (34).
Glucose and adiponectin assay
The concentrations of adiponectin in rat plasma and adiponectin in rat ovaries were measured using ELISA kits (CSB-E07271r for adiponectin, Cusabio Biotech Co., Ltd., Wuhan, China) according to the protocol. The data at 450 nm were subsequently read with a microplate reader (PT 3502 g, Beijing Potenov Technology Co., Ltd., Beijing, China). The intra-assay coefficient of variation was 4.5% and the inter-assay coefficient of variation was 7.3% in the hormone determination of adiponectin. Where ELISA experiments were made by adding 1 ml of 1\(\times\)PBS per 100 mg of tissue to make a homogenate. Glucose content was determined by the Northern Institute of Biochemistry (Beijing, China). The plasma samples were obtained from anesthetized rats, and the cell samples were obtained from purchased rats, and the ovaries were collected for primary granulosa cell culture 48 hours after the rats were acclimatized and injected with PMSG. The follicles were punctured using physical method to release the granulosa cells, which were divided into 1\(\times\)106 cell numbers into six-well plates, and the samples were collected 24 h after the addition of adipoRon, 250 µL of cell lysate RIPA were added per 1\(\times\)106 primary granulocytes.
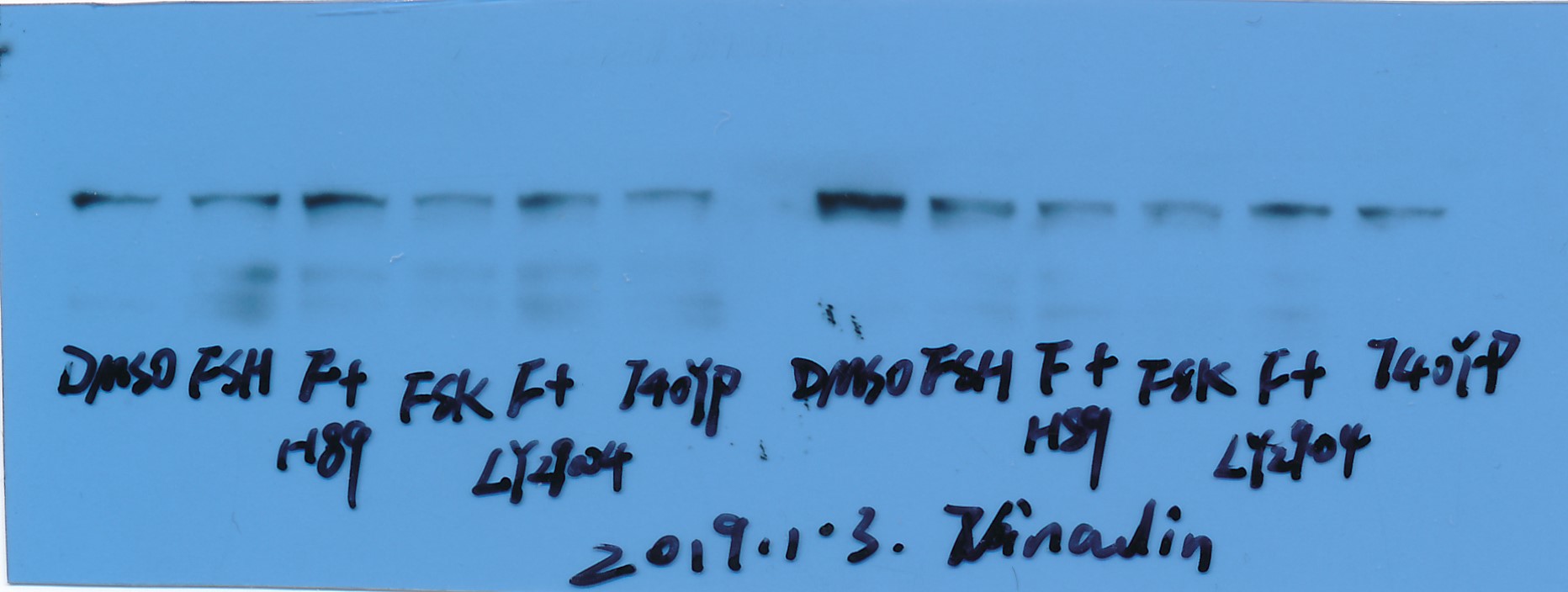
Western blot
Ovarian tissues were lysed for protein extraction using RIPA cell lysate and PMSF, followed by centrifugation (12,000 rpm, 5 min) for supernatant. All protein extractions were performed on ice, and the protein content of the supernatant (cell lysate) was determined using a BCA protein assay kit. Equal amounts of proteins were separated by SDS-PAGE (12.5%) and electro transferred onto a nitrocellulose membrane. The membrane was then blotto [Tris-buffered saline (pH 8.0) with 0.05% Tween 20 (TBS-T), 5% dehydrated nonfat milk powder] (three-phase buffered saline (pH 8.0), 0.05% Tween 20 (TBS-T), 5% dehydrated nonfat milk powder) Membranes were blocked (RT, 1 h); then washed (3\(\times\)5 min) in TBS-T with antibodies containing GLUT1 (1:1000), GLUT2 (1:1000), GLUT3 (1:1000), GLUT4 (1:1000), adiponectin (1:1000), AdipoR1 (1:1000) AdipoR2 (1:1000) and vinculin (1:5000), and incubated in blotto with HRP-conjugated secondary antibodies (1:5000, 1:5000, 1:5000, 1:5000, 1:5000, 1:5000, 1:5000, 1:5000, respectively) and washed again in TBST. Peroxidase activity was visualized with an ECL kit according to the manufacturer's instructions and protein content was determined by densitometric scanning of exposed X-ray films.
The kits and antibodies used in this experiment are listed below: PAGE Gel Fast Preparation Kit 12.5% (PG113, Shanghai Epizyme Biomedical Technology Co., Ltd), SDS-PAGE Electrophoresis Buffer (PM5060, Coolaber biotechnology, Beijing, China), TBS (T) (PM5080, Coolaber biotechnology, Beijing, China), Western transfer buffer (PM5070, Coolaber biotechnology, Beijing, China). Goat polyclonal anti-rabbit AdipoR2 (sc-46751, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Rabbit polyclonal anti-mouse AdipoR1 (sc-46748, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Goat polyclonal anti-rabbit GLUT1 (21829-1-AP, Proteintech Biotechnology, Hubei, China), Rabbit polyclonal anti-mouse GLUT2 (20436-1-AP. Proteintech Biotechnology, Hubei, China), Rabbit polyclonal anti-rabbit GLUT3 (20403-1-AP, Proteintech Biotechnology, Hubei, China), Rabbit polyclonal anti-mouse GLUT4 (sc-53566, Santa Cruz Biotechnology Santa Cruz, CA, USA), Rabbit polyclonal anti-mouse vinculin (sc-73614, Santa Cruz Biotechnology Santa Cruz, CA, USA), Rabbit polyclonal anti-mouse adiponectin (ab181281, abcam Biotechnology, abcam, Cambs, UK).
Real-time quantitative PCR
Total RNA from the ovaries of rats was extracted using the Trizol Kit (Invitrogen, Carlsbad, CA, USA) and the concentration was adjusted to 250 ng/µL. cDNA was synthesized with StarScript II RT MasterMix, RNA (1000 ng), and random primer (GenStar, Beijing, China). The 10 µL system (3 µL cDNA, 0.3 µL of forward and reverse primers (100 µg/mL), 5 µL 2× Power SYBR Green PCR master mix, and 1.4 µL ddH2O) was configured according to the FastStart DNA MasterPlast SYBR Green Kit (Roche Molecular Systems Inc., Basel, Switzerland) protocol and the primer design was shown in Table 3. The data were measured by an ABI PRISM 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). A 95℃ preheating for 10 min was followed by 40 cycles (95℃ for 30 s, 60℃ for 30 s, 72℃ for 30 s) and finally a 60–95℃ melt curve progression. The relative expression of target genes was analyzed according to the expression of internal reference β-actin. The primer design is shown in Table 1.
Table 1
Primer sequences used for mRNA RT-PCR.
| Gene Symbol | Primer Forward (5′-3′) | Primer Reverse (5′-3′) |
| Slc2a1 | AGGCCCTGGTCCTATTCCAT | CTTGTCACTTTGGCTGGCAC |
| Slc2a2 | TCATGTCGGTGGGACTTGTG | ACACGTAAGGCCCAAGGAAG |
| Slc2a3 | CAGCTCCAGCAAGCAATTCG | AGCTACCTCAAACACACCCG |
| Slc2a4 | GGCTCTGACGTAAGGATGGG | AGTGTTCCAGTCACTCGCTG |
| β-actin | GACTCGTCGTACTCCTGCTT | AAGACCTCTATGCCAACACC |
Transcriptome Analysis
Adapters and low-quality reads were removed from raw reads to get clean data. Using the previously described method, the measurement of gene expression level was obtained (35). Using the DESeq R package, the differentially expressed genes (DEGs) of two periods were examined and identified. The DEGs were identified using an adjusted P-value of 0.05, and the further implementation of enrichment analyses of DEGs using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) was performed with KOBAS software (version 3.0, accessed on 1 January 2023) (36).
Immunocytofluorescence
Granulocytes were initially inoculated into 35 mm dishes with coverslips and subsequently washed with PBS for 5 min. Following this, 4% paraformaldehyde (PFA) was added dropwise onto the cell cultures and left to incubate at room temperature for 40 min, then washed thrice with PBS for 5 min each time. Permeabilization was achieved by adding 0.25% Triton X-100 dropwise onto the cell cultures, incubating at room temperature for 10 min, and washing thrice with PBS for 5 min each. The cell slide was then treated by applying donkey serum dropwise and kept closed at room temperature for 1 h. After the removal of the donkey serum, the primary antibody solution was added dropwise onto the samples, and the antibody dilution solution was similarly applied to the negative control. Subsequently, the samples were refrigerated at 4°C overnight, brought back to room temperature the following day, and washed thrice with PBS buffer for 5 min each time. Next, the cell slide was placed in a light-proof wet box, and fluorescent secondary antibody solution was added dropwise, followed by incubation for 1 min at room temperature in the light-proof box. Following incubation, cells underwent an additional 1 h incubation at room temperature and were washed thrice with PBS for 5 min each. The staining with DAPI solution lasted for 1 min. Finally, the stained cell cultures were mounted onto slides for observation and imaging under a fluorescence microscope.
Statistical analysis
As detailed in the figure legends, results are presented as means ± SEM of at least three independent experiments. All data were subjected to one-way (repeated measure) ANOVA (Prism 9.0 statistical software; GraphPad Software, Inc., San Diego, CA). The Tukey’s test determined significant differences between treatment groups. Statistical significance was inferred at P < 0.05.
{kind=link}
{kind=link}
{kind=link}
{kind=link}