Silicon Dual-Scale Porous Membranes
‘Dual-scale’ silicon nitride membranes were purchased from SiMPore Inc. (Rochester, NY). These are commercial versions of membranes first described by Salminen et al. (2019)21. The membrane is created by patterning a microporous array atop a nanoporous silicon nitride (NPN) background. The nanoporous background has an average nanopore size of 44.6 nm, a porosity of ~ 15%, and a membrane thickness of ~ 100 nm. We patterned micropores at 5 µm in diameter to enable monocyte transmigration from the vascular to tissue sides of the hToC. We limited the microporous density to produce a ~ 1% porosity (superimposed upon the nanoporous background to give a total of ~ 16% porosity). This density of micropores is well below the 10% value shown to give monolayer stability30 and does not hinder imaging due to light scattering by micropores.
hToC Components
The top well component (top component) and bottom channel component (bottom component) of the hToC were manufactured at ALine Inc. (Signal Hill, CA) using laser cutting and lamination processes that are compatible with mid-volume production (hundreds to tens of thousands) of microfluidic components in a single production run. Parts were produced using a batch process and diced after final lamination for more reliable handling in the laboratory. The top well component contains fluidic access ports to the bottom channel that create sealed fits against P20/P200 pipette tips (VWR, 76323-390). Using pressure-sensitive adhesive (PSA), the top and bottom components were reversibly adhered to form the hToC device. Devices are free of any molded PDMS, and although they contain silicone as part of the adhesive layers, the material accounts for < 5% of the fluid-exposed surface. Thus, concerns about the loss of small molecules via absorptive loss42 are minimized in the hToC. The external surfaces of the shipped components include an additional protective layer (masking material) to maintain cleanliness and sterility during shipment and storage. The masking material is removed by the user before assembly of the components in a laminar floor hood as described in McCloskey et al. (2022)19.
Tissue Formation and Maturation
Human Tenocyte Isolation & Culture
Tenocytes were isolated from tendon tissue fragments retrieved from hand surgery procedures at the University of Rochester Medical Center under an approved Institutional Review Board (IRB) protocol (STUDY00004840). The isolated tendon tissues were immersed in alpha minimum essential medium (αMEM) supplemented with 10% Pen-Strep (10,000 U/mL penicillin/10 mg/mL streptomycin) and cut into 1-mm3 pieces using ophthalmic scissors and a gentleMACS™ Dissociator (Miltenyl Biotec, 130-093-235). After mechanical digestion, the tissue was transferred to an enzyme solution consisting of 2.5mg/ml of Collagenase D (Millipore Sigma, 11088858001), 3mg/ml of Dispase II (Millipore Sigma, D4693), and 1mg/ml of DNase (New England Biolabs, M0303) dissolved in αMEM. Tissues in enzyme solution were placed in a Roto-Therm™ Plus Incubated Rotator (Benchmark Scientific, H2024) at 37°C while rotating in combination with oscillations for an hour. αMEM supplemented with 10% Fetal Bovine Serum (FBS) and 1% Pen-Strep was added to the enzyme solution at a 2:1 ratio to inactivate the proteases in the digestive solution. The complete tissue solution was strained through a 70-micron filter, centrifuged, and resuspended in αMEM supplemented with 10% FBS, 1% Pen-Strep, and 55 µM 2-Mercaptoethanol (Thermo Fisher Scientific, 21985023). The isolated tenocytes were seeded in a T-75 flask (Corning) coated with 1 µg/cm2 Fibronectin (Sigma-Aldrich, F1056).
Cell culture was performed under standard conditions (37°C, 5% CO2, 95% humidity) with a media change after 4 days post-plating and then every other day until sub-confluence was achieved. Cells were passaged at a 1:4 split ratio using 0.25% trypsin/0.02% EDTA solution (Sigma-Aldrich, 25200056). After passage 3, the cells were cryopreserved in Recovery™ Cell Culture Freezing Medium (Thermo Fisher Scientific, 12648010) for upcoming experiments. At that point, cells were thawed and cultured in a T-175 flask under standard conditions in αMEM supplemented with 20% FBS for 24 hrs. Media was changed to αMEM supplemented with 10% FBS for 3 additional days, after which the media was changed to αMEM supplemented with 5% FBS. Tenocytes were trypsinized at day 5 after reaching ~ 80% confluency for suspension in the collagen hydrogels.
Monocytes/Macrophages
All blood samples were collected in EDTA pre-coated tubes from healthy volunteers under an IRB-approved protocol (STUDY0004777) and processed immediately after collection. PBMCs were isolated from whole blood by density separation over a solution of 1-Step™ Polymorph (Accurate Chemical & Scientific Co., AN221725) at 500×g for 30 min without brakes. The white buffy PBMC layer was then washed twice in Wash buffer (HBSS without Ca2+ and Mg2+ with 10mM HEPES and 5mg/ml BSA) and centrifuged at 350×g for 7 min without brakes to remove platelets. Red blood cells were lysed by exposing the washed PBMC layer to 1/6x PBS for 1 min, followed by 4x PBS for 1 min, at a 3:1 ratio, and spinning at 350×g for 7 min. PBMCs were resuspended in Wash buffer and spun down at 350×g for 7 min to remove any remaining PBS. PBMCs were resuspended in an Isolation buffer (1XDPBS without Ca2+ and Mg2+ with 2mM EDTA and 1 mg/ml BSA). CD14+ Monocytes were isolated from PBMCs by positive magnetic enrichment using the QuadroMACS Starting Kit (LS) (Miltenyi Cat# 130-091-051) using the manufacturer's protocol.
Cell-laden hydrogel
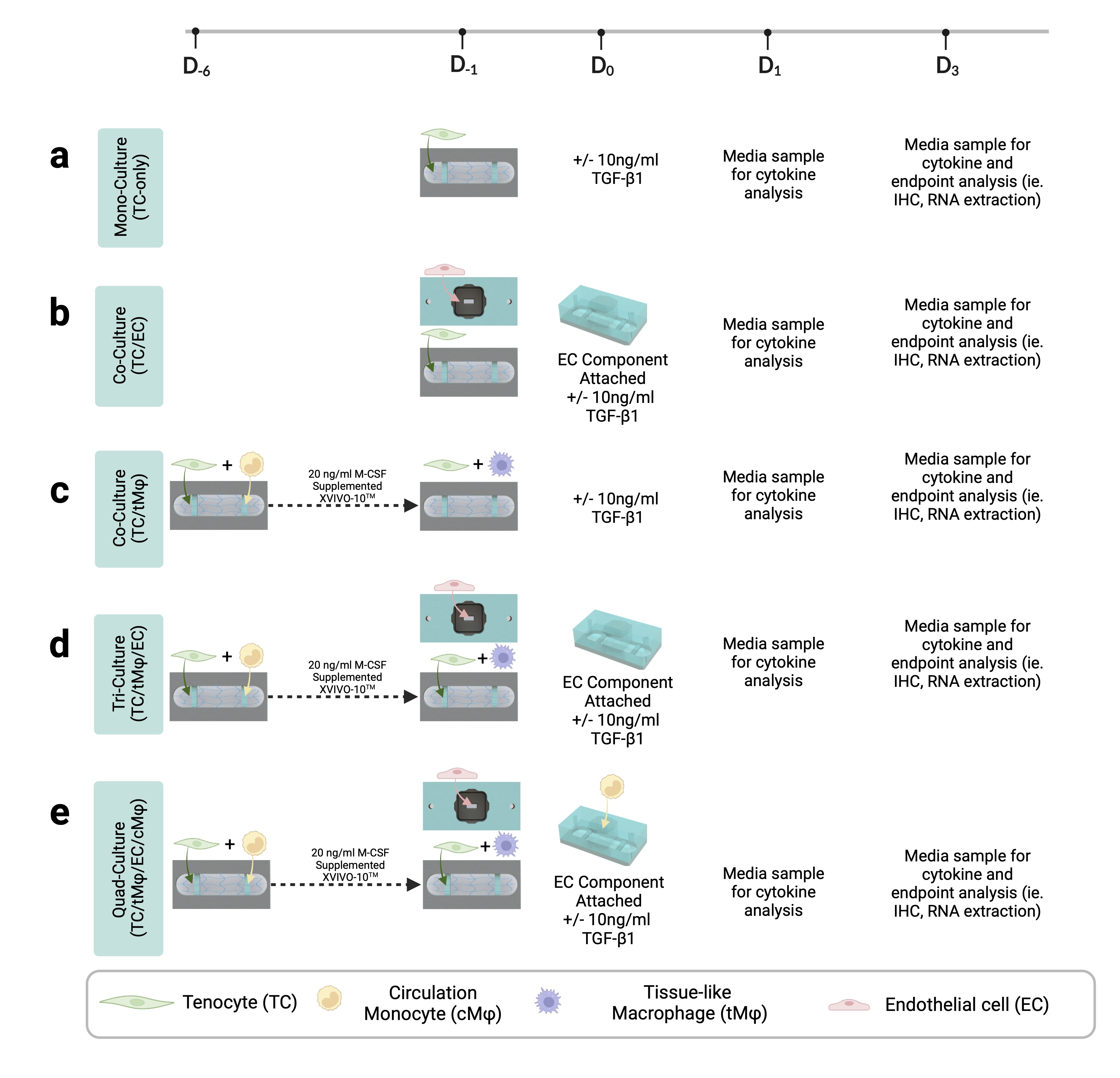
For TC-hydrogel monocultures, P3-P6 tenocytes were suspended at 500,000 cells/ml in collagen hydrogel (TeleCol®-3, Advanced Biomatrix, 5026). The cell suspension was introduced into the collagen stock solution at a 1:18 ratio to achieve a final hydrogel concentration of 2.6 mg/ml. The hydrogel mixture was pipetted into the bottom channel and supplemented with 200 µl of X-VIVO™ 10 media. At D0, the media in the reservoir was replaced with ± TGF-β1 supplemented X-VIVO™ 10 (10 ng/ml TGF-β1 (R&D Systems, 240-B-002), 20 µg/ml plasminogen (Haematologic Technologies, HCPG-0130), and 50 ng/ml of tPA (Fisher Scientific, NBP25955350).
For TC/tMϕ co-cultures, freshly isolated monocytes were suspended in TeleCol®-3 with tenocytes at a ratio of 1:7. The hydrogel was pipetted into the bottom channel and 200 µl of X-VIVO™ 10 media supplemented with 20 ng/ml M-CSF (PeproTech, 300 − 25) was added to induce monocyte polarization into naïve macrophages. M-CSF supplemented media was entirely replaced on D− 4 and D− 2 and ½ replaced on D− 5 and D− 3.
Endothelial Cells
Pooled Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from LONZA (C2519A) and maintained in a T-75 flask at 37°C, 5% CO2, 95% humidity in EGM-2™ media (LONZA, CC-3162). HUVECs were used between passage number 3–7 as recommended by the supplier and cultured according to the supplier’s protocol.
Quad-Culture hToC Assembly
The hToC top component was assembled as described above. To prevent bubble formation at the interface of the trench of the porous membrane and the tissue hydrogel in the bottom channel, the membrane trench was backfilled with collagen solution and was then placed in the incubator at 37°C, 5% CO2, 95% humidity for 20 min to allow the collagen hydrogel to crosslink. After 20 min, the top components were removed from the incubator, and the reservoirs were attached and filled with 225 µl of EGMTM-2 media. The back-filled top components were placed onto the media-filled reservoirs to prevent the collagen hydrogel in the trench from drying out. The well-side of the porous membrane was then coated with 0.17 mg/ml fibronectin (Sigma, F1141) for 1 hr at room temperature to facilitate cell adhesion. Expanded HUVECs were plated into the 100 µl top component well at a 40,000 cell/cm2 density. Cells were allowed to settle for 3 hours before rinsing with EGMTM-2 media to remove non-adherent cells. HUVECs were incubated for 24 hours in the top component alone before combining with the bottom component containing the TC/ tMϕ hydrogel. EGMTM-2 media was replaced with X-VIVO™ 10 media in the top component well after assembly to establish serum-free conditions during the experimental timeline.
For tri- and quad-culture assembly, the top component is adhered to the bottom channel to form a tendon-endothelial barrier. Prior to assembly, the bottom channel cultures must be ready for assembly (24-hr culture for TC only and 6-day culture for TC/tMϕ co-cultures), the ECs in the top component must have formed a confluent monolayer (expected after 24-hrs), and CD14+ monocytes freshly isolated. After full assembly of the hToC, the bottom channel was filled with 100 µl of X-VIVO™ 10 media ± TGF-β1. Freshly isolated CD14+ monocytes suspended at 100,000 cells/ml in 100 µl of X-VIVO™ 10 media were added to the top component well containing the EC monolayer. For all rapamycin experiments in this study, a 10 ng/ml rapamycin solution was added to the EC well at D0. The fully assembled hToC was then placed in an incubator and cultured at standard conditions. Culture supernatants from the top well and bottom channel were sampled from 24- and 72-hour devices for cytokine analysis. At the end of the experiments, EC monolayers and hydrogels were fixed or processed for immunofluorescence or alternative downstream assays at 24 and 72 hours, respectively.
Live-Stain Imaging and Quantification of Macrophage Transmigration in hToC
TC, tMϕ, and cMϕ were live-stained using the ViaFluor SE Cell Proliferation Kit (Biotium, 30068-T) before seeding into the hToC and imaged at 3-, 24- or 72- hrs, respectively. For each image, a z-stack was taken every 10 minutes over 45 minutes, starting ~ 50 µm on top of the porous membrane and 600 µm under the porous membrane to capture cMϕ migration through the EC monolayer and into the bottom channel compartment. The hToC was placed on an incubation stage at standard conditions for the entire imaging session to prevent cell death during imaging.
Luminex Multiplex Immunoassay
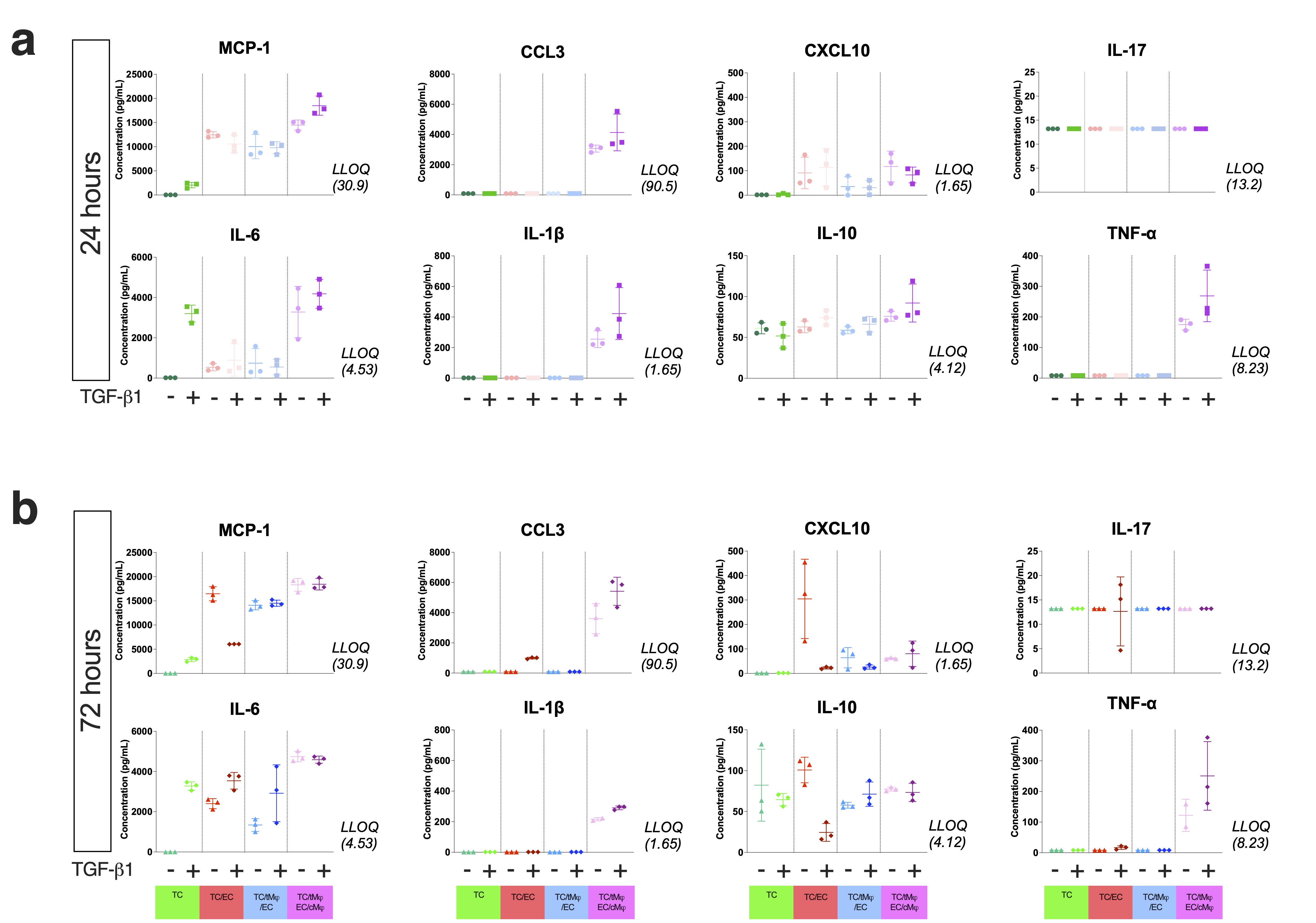
Human Luminex Discovery Assay (R&D Systems) was used for the quantification of selected human cytokines and chemokines: MCP-1, CCL3, CXCL10, IL-1β, IL-6, IL-10, IL-17, and TNF-α in cell supernatants collected from the top well and bottom channel (~ 110µl) and frozen immediately at -80°C. The measurements were performed according to the manufacturer’s specifications using a Luminex 200 Instrument. The experiments were performed with at least three biological replicates per condition, each performed in duplicate.
Immunostaining and Fluorescence Microscopy
Tendon constructs were rinsed with 1× PBS, fixed in 4% Paraformaldehyde (PFA) for 10 min, and then washed 2 × 5 min in PBS. Cells were then permeabilized with 0.5% Triton X-100 (Amresco, 0694) for 10 min and rinsed. The constructs were blocked in 1% Bovine Serum Albumin (BSA) (Cell Signaling Technology, 9998) solution for 30 min at Room Temperature (RT) and incubated with the primary antibody solutions (Supplemental Table) diluted in 0.1% BSA or PBS overnight at 4°C. The respective secondary antibody (Supplemental Table 2) used at the manufacturer’s recommended dilution was diluted in 0.1% BSA or PBS and incubated for 1 hr at RT. Hoechst stain was then diluted in PBS and incubated for 10 min at RT. Alternatively, hydrogels were fixed in 4% PFA, dehydrated, removed from the channel and embedded in paraffin. The blocks were sectioned at 10 µm thickness. For IHC, deparaffinized and rehydrated sections were incubated in 10 mM sodium Citrate (pH 6.0) for 1 hr at 65°C for antigen retrieval. Slides were rinsed with ddH2O for 5 min followed by 3 × 4 min washes in PBS and 2 × 4 min washes in PBST (PBS + 0.1% Tween-20). Slides were blocked for 1 hr in 5% BSA diluted in PBST and incubated with primary antibodies (Supplemental Table) at 4°C overnight. After primary antibody incubation, the slides were washed 3 × 10 min in PBS and then incubated with respective secondary antibodies diluted in 5% BSA diluted in PBST. Slides were then rinsed 3 × 5 min in PBST, followed by a 2 × 5 min wash in PBS. Sections were cured with ProLong™ Diamond Antifade Mountant with DAPI (Thermo Fisher, P36962) overnight at RT in the dark. Clear nail polish was used as a sealant the following day, and slides were stored at 4°C until imaged.
For ICAM-1 and VCAM-1 expression in EC monolayers, live stains were used (Supplemental Table 1). This required adding the primary antibodies diluted in X-VIVO™ 10 media and incubating at standard conditions for 15 min before fixing. Cells were then blocked with blocking buffer (5% BSA + 0.1% Triton X-100) and incubated at RT for 10 min. For ICAM-1 and VCAM-1, secondary antibodies and Hoechst stain were added in blocking buffer and incubated for 1 hr at RT. In the case of VE-Cadherin and PECAM-1 staining, primary antibodies were diluted in blocking buffer and incubated for 1 hr at RT after the blocking step. After rinsing the well, the secondary antibody + Hoechst stain diluted in blocking buffer were added and incubated for 1 hr at RT. All samples were stored in a humidified chamber at 4°C and protected from light until imaging. All images were acquired using a Dragonfly Spinning Disk Confocal System (Andor, Belfast, UK) at the University of Rochester High Content Imaging Core.
RNA Extraction from Collagen Construct and Bulk RNA-Seq
The cell-laden hydrogels (n = 6 samples per group, with each sample consisting of 2 pooled hydrogels) were harvested from the hToC bottom compartment immediately following the experiment. The hydrogel was digested to release the cells by adding 500 µl of 1 mg/ml (215 units/mg) of collagenase I (Thermo Fisher, 17018029) diluted in X-VIVO™ 10 medium and stored at standard conditions for 45 min. Total RNA was isolated from released cells using the RNeasy Plus Micro Kit (Qiagen, 74134) per manufacturer recommendations. To isolate RNA from the EC monolayer, 100 µl of Lysis Buffer was added to the top component well for 10 min. The EC lysate was immediately processed following the manufacturer's protocol. The RNA concentration was determined via NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE), and RNA quality was assessed with the Agilent Bioanalyzer (Agilent, Santa Clara, CA). Messenger RNA isolation and next-generation RNA sequencing analysis were performed by the University of Rochester Genomics Core.
Bioinformatics and statistical analysis of DEGs
All bioinformatic analysis was performed with original RStudio scripts (R Core Team (2022). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.URL https://www.R-project.org/). Using DESeq2, gene expressions were compared between the human (uninjured and tenolysis from Zheng et al16) and hToC samples (vascular and tissue separately). To correct for differences attributed to batch effects, all count data was batch adjusted using ComBat-seq. The Wald test was used to generate P values and log2(fold changes). Genes with P < 0.05 and absolute log2(fold changes) > 1 were selected as differentially expressed genes for each comparison. Heatmaps, dot plots, and enrichment maps were performed using the Complex Heatmap Visualization and Enrichplot packages. Pathway analysis for Gene Ontology (GO), Kyoto encyclopedia of genes and genomes (KEGG) and Reactome were performed using their respective homo sapien database in RStudio. The differentially expressed genes obtained from RNA sequencing were ranked from highest to lowest expression and uploaded to GSEA software.
{kind=link}
{kind=link}