Background information regarding the subject’s natural history is collected using a REDCap© survey (Copyright 2006–2013 Vanderbilt University. All rights reserved) [16]. Body weight and height are measured at each study visit. Serum laboratory analysis is conducted to establish a metabolic baseline: comprehensive metabolic panel, complete blood count, ketones (beta-hydroxybutyrate and acetoacetate), and lipid panel. Electroencephalography (EEG), event-related potential recordings (ERP) and gait analysis using the ProtoKinetics Zeno™ Walkway are performed at each study visit (Figure 1). The utility of ERP and gait analysis to predict changes in patient function in an Angelman population has not been clearly established, but their appropriateness in similar populations has been detailed elsewhere [17–18]. Questionnaires completed during the study include the Vineland™–3, Developmental Behavioral Checklist (Pearson Education, Inc.), which has been used previously in studies of patients with AS [19]. Dietary intake, seizure frequency, sleep and GI health are evaluated throughout the protocol through at-home monitoring. Data collected during home monitoring is recorded on tablet devices provided to the parents/caregivers upon enrollment, preloaded with necessary tracking applications. Families are provided an EarlySense® monitor with a written guidance for home sleep monitoring. External sleep monitors are important in an AS population as wearable devices are typically removed by the patient. The monitors have not been studied with patients with AS, however their validity has been demonstrated in the general population [20]. Urinary ketones are measured by the patient’s caregivers throughout the protocol, using Ketostix which are a validated measure of urine acetoacetate [21–22].
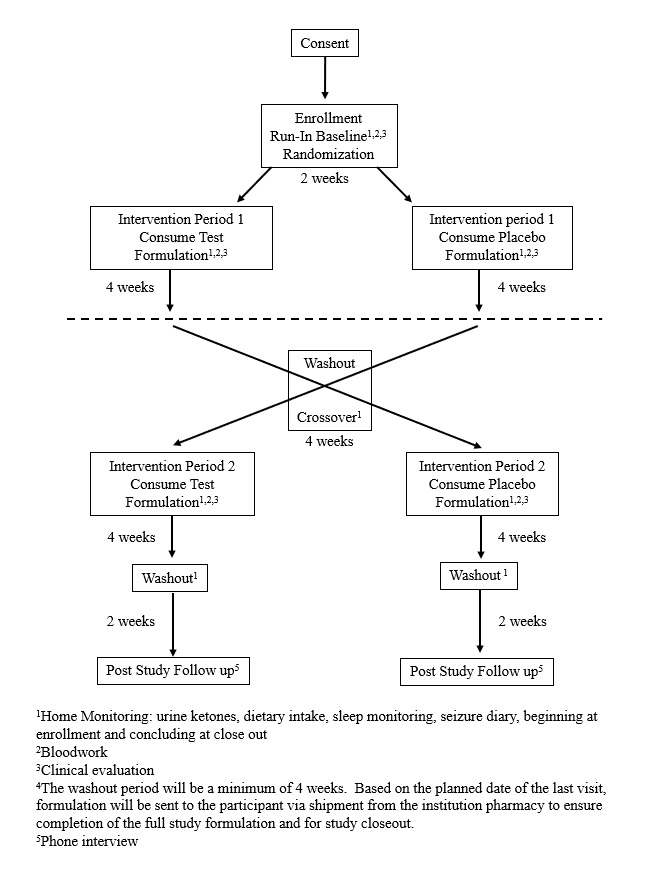
Tolerability is determined at the end of each four-week intervention period. Protocol compliance is monitored during both investigational and placebo intervention periods by recording the amount of formulation consumed. Formulation acceptability is assessed at the end of each intervention period by a parent questionnaire evaluating convenience, taste and degree of acceptance of the nutritional formulation on a ten-point Likert scale. Similar ranking systems to assess formulation acceptability have been used with other nutritional interventions in epilepsy populations [22–25].
Safety is assessed throughout the entire protocol period through (1) adverse event reporting and (2) assessment of clinical parameters collected throughout the protocol including anthropometrics, blood metabolism profile, dietary intake, seizure frequency, EEG, ERP, mobility, GI function, and sleep habits. In this population, the potential for negative effects of intervention must be closely monitored as patients are nonverbal. Additional File 4 contains details on collecting, assessing, reporting, and managing solicited and spontaneously reported adverse events.
Investigational and placebo formulations are administered with the serving size based on subject body weight. The investigational formulation provides beta-hydroxybutyrate, 2g of carbohydrate, 1g of protein, and 9g of fat, plus minerals, per 100kcal. The placebo is matched for mineral content only. The formula is given orally as food or beverage three times per day.
Families will be encouraged to continue participation in the trial through weekly telephonic interviews. Data will be collected through three primary interfaces. Background information regarding the patient’s natural history will be collected by the clinical staff using the REDCap survey. REDCap© is a secure web application that manages the trial database (21 CFR Part 11 and HIPAA compliant). Clinical visits will use either paper or electronic medical records. Data collected during home monitoring will be recorded using a tablet provided to the patient upon enrollment. The tablet is preloaded with the applications necessary to record all data during home monitoring. All data collected, regardless of patient completion status, will be shared with patients upon conclusion of the trial (last patient last visit) and assessment of the data. Additional File 4 contains details on data management and collection procedures.
Statistical Considerations
The primary outcome of the trial is to assess the tolerability of a nutritional formulation containing BHB in patients with AS. Tolerability is demonstrated through patient compliance with the protocol as determined by the amount of formulation consumed as compared to the amount prescribed and is assessed at the end of each intervention period. Premature discontinuation of the nutritional formulation by the family is also deemed as potential intolerability. Tolerability is also assessed at the end of each intervention period through parental questionnaire. These outcome measures were chosen as this is a vulnerable, nonverbal population with limited ability to communicate and therefore determining if the formulation is suitable and well tolerated is an appropriate first step.
The secondary outcomes include assessment of ketosis when consuming the nutritional formulation and safety of the nutritional formulation in patients with AS. Ketosis is evaluated daily through urine testing as well as blood analysis at the end of each intervention period. The degree and timing of ketosis may be clinically relevant to demonstrating better patient nutrition as nutritional ketosis has been related to better patient outcomes as described in the background of this manuscript. Safety is evaluated by changes in motor function, cognitive function, GI tolerance, sleep and seizures, along with changes in height, weight and blood metabolic panels as assessed at the end of each intervention period. In addition, adverse event reporting is monitored in real time by the study coordinator.
No studies testing ketone supplementation in patients with AS exist. Therefore, sample size estimation is based on measures of ketosis. It is assumed from murine pre-clinical investigations, non-AS clinical studies using MCT and/or BHB, and low carbohydrate dietary interventions, that the minimal change of the secondary outcome, degree of ketosis, from the baseline to the end of the 4th week of the investigational formulation intervention period will be 100% change from baseline (baseline for standard diet is typically <0.5mMol/L of d-BHB in blood, or < 5mg/dL acetoacetate in urine). The acetoacetate test used is Ketostix (Bayer Corp. Diagnostics Division, Tarrytown, New York) which exhibits a sensitivity of 78% and a specificity of 96% when compared to a serum standard of 14.4 ml/dL. Considering a 25% drop out rate, an estimate of 25 subjects need to be screened to achieve 15 completers.
Descriptive statistics, such as mean and standard deviation, are used to describe continuous variables, and frequency for the categorical variables. Means are compared using a paired two-sample t-test. Statistical significance for all tests is p<0.05. Multiple comparisons can be made including: a) each subject to serve as his or her own control, comparing investigational formulation to placebo formulation, as well as to baseline, b) each dietary background group considered in aggregate, comparing investigational formulation to placebo formulation, as well as to baseline, and c) all subjects considered in aggregate, comparing investigational formulation to placebo formulation, as well as to baseline.
Any patient that receives study formulation for any amount of time will be included in the intent-to-treat population for study analyses. Missing data points in this population will be addressed in discussion of any published results. Data from protocol compliant participants are the defined population for efficacy subset analyses; any participant who received the both protocol-required formulations and all required clinical assessments are considered protocol compliant. Such an efficacy subset analysis will be the primary approach for determining if trial endpoints are met. Those patients that complete the baseline visit, but do not start Intervention Period One and are withdrawn from the study, will be used for natural history reportable data only but will not be included in analyses for determining whether the outcome measures of the trial have been met.
Discontinuation/Withdrawal from Study Participation
Participation in this study is voluntary. The parent or guardian of a participant may withdraw consent to participate in the study at any time for any reason they deem necessary. The administration of the study formulation may be discontinued abruptly and does not require any further medical intervention and no alternate therapy will be offered.
Data Monitoring
In order to ensure the safety of the study participants, members of a Data Safety Monitoring Board (DSMB) have been appointed by the principal investigator and Vanderbilt University. The board consist of a chairperson, biostatistician, and three independent reviewers with expertise in the research field. Each member of the board meets these minimum qualifications including: 1) expertise in the field, 2) experience in the conduct of clinical trials and statistical knowledge, 3) independence from the direct management of the clinical trial, and 4) no conflict of interest. The chairperson is responsible for overseeing the meetings, developing the agenda, summarizing the meeting, and is the contact person for the DSMB. The DSMB meets 3–4 times per year, such schedule to be determined by the chairperson. Additional details on the charter of the DSMB can be had by contacting the Vanderbilt University Medical Center Human Research Protections Program at www.vumc.org/irb.
Ethical Considerations
This study is to be conducted according to US and international standards of Good Clinical Practice (FDA Title 21 part 312 and International Conference on Harmonization guidelines), applicable government regulations and institutional research policies and procedures. This protocol and any amendments will be submitted to the Vanderbilt University institutional review board (IRB) in agreement with local legal prescriptions, for formal approval of the study conduct. The DSMB will work in conjunction with the principal investigator and IRB to determine if a protocol amendment warrants additional patient consent or other communication to the enrolled patients and their families. The decision of the IRB concerning the conduct of the study will be made in writing to the investigator before commencement of this study. Protection of participant confidentiality is described further in Additional File 4.
Participants may be able to claim compensation for injury caused by participation in this clinical trial. Participants who sustain injury and wish to make a claim for compensation should do so in writing in the first instance to the Principal Investigator, who will pass the claim to the Sponsor’s Insurers, via the Sponsor’s office.
{kind=link}