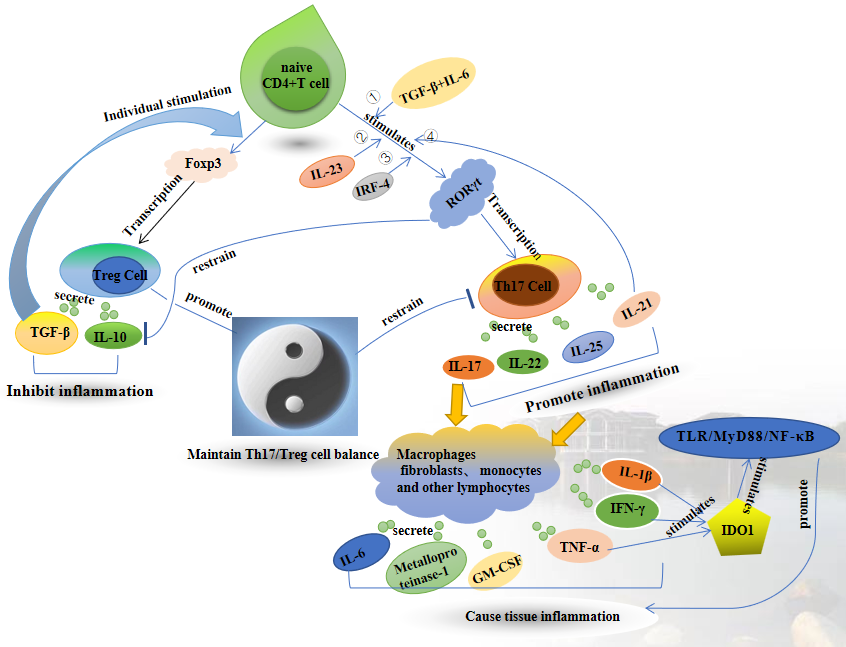
The immune imbalance of Th17/Treg is one of the important contributing factors for promoting inflammatory response and inducing autoimmune diseases. The IL-6-JAK-STAT3 pathway upregulates the expression of transcription factors RORγt and RORα, thereby facilitating the differentiation of Th17 cells. These Th17 cells subsequently release characteristic cytokines, including IL-17A, IL-17F, IL-21, IL-22, and IL-23[18]. IL-17 plays a crucial role in the initiation of inflammation by stimulating the synthesis of IL-6 and IL-8 via the mitogen-activated protein kinase (MAPK) pathway, thereby promoting the recruitment of neutrophils to the site of inflammation and triggering the proliferation of T cells. Additionally, IL-17 also promotes the expression of certain pro-inflammatory molecules such as inducible nitric oxide synthase (iNOS) and IL-1β[19]. Treg cells possess immunomodulatory capabilities, enabling them to suppress immune responses triggered by diverse antigens and promote immune tolerance. In the presence of inflammation, activation of dendritic cells, monocytes, and macrophages prompts the rapid migration of Treg cells to the inflammatory site, effectively impeding the initiation and progression of inflammation at the site, thereby preserving immune homeostasis.
When the gut is stimulated to damage epithelial cells, inflammatory factors stimulate dendritic cells to produce IL-6 and up-regulate transcription factor RORγt, thus promoting Th17 cell differentiation and producing IL-17[20]. At the same time, it also promotes the recruitment of neutrophils into the intestine, stimulates fibroblasts, macrophages, etc., and then induces the production of a variety of pro-inflammatory cytokines, resulting in intestinal inflammation. Continuous inflammatory stimulation leads to a decrease in the number of Treg cells and a weakening of their function. At this time, CD4 + T cells tend to differentiate into Th17 cells, and the pro-inflammatory factors secreted by Th17 cells rapidly increase, leading to further aggravation of intestinal mucosal inflammation [21]. The secretion of IL-10 by Treg cells has the ability to impede the synthesis of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, thereby exerting an inhibitory influence on T-cell-mediated immune responses[22]. The functional opposition between Th17 and Treg cells is intricately linked to the development of chronic inflammatory responses, malignant tumors, and various autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, and inflammatory bowel disease [23–27]. UC is a chronic non-specific inflammatory disease of the intestine, wherein the dysfunction of intestinal mucosal immune system plays a crucial role in the pathogenesis of UC. This dysfunction encompasses abnormalities in both innate and adaptive immunity within the intestinal mucosa. At present, studies on congenital immunity mainly include the integrity of intestinal epithelial barrier and autophagy. Conversely, studies pertaining to adaptive immunity primarily concentrate on the inflammatory response mediated by B cells and T cells within the lamina propria of the intestinal mucosa[28, 29]. Th17 and Treg cells are integral components of the intestinal mucosal immune system, exerting significant influence.When the gut is stimulated to damage epithelial cells, inflammatory factors stimulate dendritic cells to produce IL-6 and up-regulate the expression of transcription factor RORγt, thus facilitating the differentiation of Th17 cells and producing IL-17[30]. Simultaneously, it has the potential to facilitate the recruitment of neutrophils to the intestinal region, activate fibroblasts, macrophages, and other immune cells, as well as trigger the synthesis of various pro-inflammatory cytokines, culminating in the onset of intestinal inflammation. Prolonged exposure to inflammatory stimuli results in a reduction in the population of Treg cells and a decline in their regulatory function. At this time, CD4+ T cells exhibit a propensity to differentiate into Th17 cells, which in turn secrete heightened levels of pro-inflammatory mediators, thereby exacerbating the inflammatory response within the intestinal mucosa [21]. The gut's innate immune response facilitates the generation of Treg cells while restraining the excessive inflammatory reaction of effector T cells. Consequently, preserving the equilibrium between Th17 and Treg cells emerges as a crucial strategy in averting the onset of UC.
The colitis model of DSS induced intestinal epithelial injury is one of the most widely used UC models [31]. In this study, it was found that during the construction of UC model, QFG can inhibit weight loss in UC mice, relieve diarrhea, fecal occult blood, and DAI index. It alleviated the shortening of colon length induced by DSS and alleviated the pathological injury of colon tissue. These results suggest that QFG can improve the overall well-being of mice and effectively resist the intestinal damage induced by DSS in UC mice.
Given that DSS has been observed to induce notable colon inflammation in mice with UC, it is noteworthy that the immune cells infiltrating the inflamed tissues release pro-inflammatory cytokines such as IL-1β, TNF-α, IFN-γ, IL-6, among others. It has been established that the expression levels of these factors exhibit a positive correlation with the severity of UC, thus rendering them suitable indicators for assessing the severity of this condition [32]. Therefore, in this study, we detected the levels of IL-1β, TNF-α, IFN-γ, IL-6 and other cytokines in serum, spleen and colon tissues of mice, so as to determine whether QFG can inhibit colon inflammation in UC mice. In addition, during the occurrence and development of UC, IDO1 is also significantly elevated. This enzyme is the first rate-limiting catalytic enzyme of tryptophan along the kynuridine metabolic pathway and is widely expressed throughout the body.
Furthermore, in the context of ulcerative colitis (UC), there is a notable increase in the expression of indoleamine 2,3-dioxygenase 1 (IDO1). This particular enzyme serves as the primary regulatory catalyst within the kynuridine metabolic pathway for tryptophan, and exhibits widespread distribution across various bodily tissues. Studies have confirmed that augmented levels of inflammatory cytokines such as IL-1β, IFN-γ and TNF-α, elicit a notable upregulation of IDO1 [33, 34], especially in colon tissue. In addition, increased expression of IDO1 can promote the occurrence and development of inflammation through mediating TLR4/MyD88/NF-κB signaling [35, 36], which is an important inflammatory signal in the development of UC [37] and an important indicator to evaluate the prognosis of UC inflammation. In this study, the results showed that the expression of cytokines such as IL-1β, TNF-α, IFN-γ, IL-6 and IDO1 in UC mice was significantly increased compared with that in the Control group, indicating that the DSS induced UC model had a significant inflammatory response. However, QFG decreased the expression of IL-1β, TNF-α, IFN-γ, IL-6, IDO1 in mice, indicating that QFG inhibited colon inflammation in UC mice.
In order to explore whether the mechanism of QFG inhibiting colon inflammation in UC mice is related to the regulation of Th17/Treg cell balance, we examined the changes of cytokines related to Th17 and Treg cells. Inflammation stimulates the body. First, Treg cells play a role in intestinal innate immunity and accelerate Treg cell differentiation to inhibit T cell-mediated inflammatory response. The results of this study showed that after DSS administration, the cytokines associated with Treg cells in mice were slightly increased, which may be related to the innate immunity of Treg cells. With the continuous stimulation of inflammation, the number of Treg cells decreased and their function weakened, while Th17 cells took advantage of differentiation and secreted pro-inflammatory factors rapidly increased, leading to further aggravation of intestinal mucosal inflammation. This is consistent with the results of this study, which showed that after DSS administration, the cytokines related to Th17 cells in mice were significantly increased, while the cytokines related to Treg cells, TGF-β and IL-10, were slightly increased. After QFG treatment, Th17-related cytokines IL-17, IL-21, IL-22 and IL-25 were significantly decreased, while Treg-related cytokines TGF-β and IL-10 were significantly increased, restoring the balance of Th17/Treg cells. This may be related to QFG inhibiting the differentiation of Th17 cells and promoting the differentiation of Treg cells. Therefore, we further examined the changes of transcription factor RORγt in Th17 cells and Foxp3 in Treg cells. The results showed that RORγt was significantly increased and Foxp3 was slightly increased after DSS treatment. After QFG treatment, RORγt was significantly decreased and Foxp3 was significantly increased, which was consistent with the changes of cytokines related to Th17 and Treg cells. This suggests that RORγt and Foxp3 affect the changes of Th17 and Treg cell related cytokines. Therefore, QFG may inhibit the transcription of RORγt and thus inhibit the differentiation of Th17 cells, and promote the up-regulation of transcription factor Foxp3 to increase the differentiation of Treg cells and maintain the Th17/Treg cell balance. Thus, the intestinal inflammatory response of UC mice induced by DSS was inhibited.
{kind=link}