Ethics statement
The ethical committee of Yunnan Animal Science and Veterinary Institute (Kunming city of Yunnan province, China) has approved all experiments including animal usage in this study (201909006). In addition, during the whole experiment, the authors strictly complied with Regulations on the Administration of Laboratory Animals (Order-No.2 of the State Science and Technology Commission of the people's Republic of China, 1988) and Regulations on the Administration of Experimental Animals of Yunnan Province (the Standing Committee of Yunnan Provincial People's Congress 2007.10). We confirmed that all authors complied with the ARRIVE guidelines.
Chemicals and reagents
Unless otherwise mentioned, the chemicals, reagents, and kits have been purchased from Sigma-Chemical Company (St. Louis, Mo, United States). The Andromed extender was purchased from Minitüb GmbH (Hauptstrasse 41, 84184 Tiefenbach, Germany).
Animals and management
In this study, the semen used was collected from a newly developed breed-Yunshang black goats. To collect semen, 20 bucks (2–3 years old) were used during September of 2019 (their reproductive season). Routine anthelmintic handling and vaccination against rabies and tetanus were conducted. The bucks were raised under the standardized conditions of feeding, lodging and light. The daily diet consisted of 29.5% maize, 23% soybean, 1.5% calcium monophosphate, 1% premis, 0.5% sodium-bicarbonate, 0.5% NaCl, 19% broad bean-bran, 10% alfalfa-Grass, and 15% corn-silage. The bucks had free access to salt and drink.
Semen collection, dilution, and motility assessment
In this study, semen was collected using artificial vagina and directly transported to the laboratory within 10 minutes. Two successive ejaculates of one buck obtained over a 10 min period were pooled for its semen quality analysis. Instantly after collecting, we counted volume of semen and observed semen color. Mass motility was first assessed by observing the wave motion pattern of fresh undiluted semen 76, 77. However, the assessment of mass motility is subjectively carried out on the basis of the experience and knowledge of the technicians, so it is only a rough assessment. Concentrations of sperm were analyzed using Nucleo-Counter ® SP 100™ (Chemo-Metic AS, Allerød, Denmark). Following the initially assessment, quality of the used ejaculates satisfied with the criteria in the experiments were as follows: mass motility: ≥3.0; sperm concentration: ≥2500×106 sperm/mL; normal morphology: ≥75%.
After the above mass motility assessment, the motility of sperm was analyzed using a computer-assisted sperm CASA system installed with the Sperm Class Analyzer (SCA) software (SCA Evolution; Microptic, Barcelona, Spain). A specific program in this software is designed for the evaluation of goat sperm. The detailed parameter setting for this program was as follows: Calibration name, 10×; Calibration value (μm/pixel), 0.475323; Capture method, Ph-; Grid distance (μm), 100; Analysis timeout, 15; Box size, 152; Frame rate (fps), 25; number of images, 25; Resolution, Low; Style, automatic; Minimum Area, 3μm2; Maximum Area, 70μm2; Drifting (μm/s), 0; Static (μm/s) <10; slow-medium velocity (μm/s), 45; Rapid velocity (μm/s), 75; progressive motility (STR>), 80; connectivity (pixels), 12; VAP points (pixels), 5; VCL/VAP, VCL.
When the motility was examined, the collected semen samples were diluted using the Andromed extender to a final concentration of 20×106 sperm/mL. 10μl drop of sperm solution was placed on a slide and covered using a cover slip (18mm × 18mm). Initially, the heated plate (38°C) with a magnification of 100× have been installed on a phase-contrast microscope (Nikon, ECLIPSE-E200, Japan), and the progressive motility (PM, %) values were analyzed. Ten fields per drop including a total of 500 sperm has been recorded for every sample. Based on the obtained sperm motility values, the used bucks were separated into two groups with a higher (≥75%) or lower motility (≤65%).
When performing the proteomic analysis of goat seminal plasma, there are 5 bucks with higher or lower motility in each group. However, when performing the metabolomic analysis of seminal plasma, there are 10 bucks with higher or lower motility in each group. The semen from these bucks used were analyzed separately and not pooled during this whole experiment.
Sperm plasma membrane and acrosome assessment
The hypo-osmotic swelling test (HOST) has been used to test the integrity of sperm plasma membrane as described in a previous study 78. In brief, 20 µL of semen was incubated in 200 µL of the hypo-osmotic solution (9 g/l fructose and 4.9 g/l sodium citrate, 100 mOsm/kg) at 37 oC for 60 minutes. Then, 10 µL of solution was mounted on a microscope slide and covered using a cover slip. A total of 200 sperm were assessed in each time. Sperm with visible coiling tails were counted under the phase contrast microscope with a magnification of 400× for each sample.
FITC-PSA staining together with flow cytometry was used to assess the acrosome status of goat sperm 77. In brief, semen was diluted using the TALP buffer to a fixed concentration of 10×106 sperm/mL. Then, 200 μL of the above sample was stained using 50μL propidium-iodide (PI) (50μg/mL) and 0.5μL FITC PSA (2mg/mL), followed by incubation in a dark and humid environment for 15 minutes at 37°C. Finally, the percentages of FITC-PSA and PI stained sperm were analyzed by flow cytometry. The concentration of alive sperm with intact acrosome and plasma membrane were identified as PI and FITC-PSA negative.
A FacStar-plus flow cytometer (FAC SCalibur, Becton-Dickinson and Co., Franklin Lakes, NJ, USA) was used to perform the flow cytometry analysis. The green fluorescence emitted from FITC-PSA were detected on the FL1 photodetector (530/30BP-filter). The red fluorescence generated from PI was observed on the FL2 photodetector (670LP-filter). The Ar ion blue laser was used to excite those fluorochromes (488 nm). The fluorescence information was shown in the logarithmic mode using the Cell-Quest Pro-3.1 program (BD-Biosciences). According to the guideline of International Society for the Advancement of Cytometry (ISAC), the data was obtained from 100,000 events for further study using the Cell-Quest program (Becton Dickinson).
Seminal plasma exaction and purification
The seminal plasma exaction process was defined in a previous report 79. In brief, following semen collection, seminal plasma was extracted separately from sperm cells via centrifugation at 10,000 × g for 10 minutes in a microfuge at 4oC. Then, the supernatants were gently collected and centrifuged again at the same condition. The collected seminal plasma was further filtered via a 0.22 μm Millipore filter (Millipore). The seminal plasma samples were preserved at -80 oC for the proteomics and metabolomic analysis.
Protein extraction and trypsin digestion
The protein extraction process has been described in a previous study 32. In brief, before the extraction of total proteins in seminal plasma, all samples were initially sonicated for three times using ice, applying the highly intensity ultrasonic-processor (Scientz) in the lysis buffer (8M urea, 1% protease inhibitor cocktail). The supernatants were collected after centrifugation at 12,000 g at 4 oC for 10 minutes, and the protein concentrations were measured using the BCA kit as instructed by the manufacturer.
The protein mixture was reduced by 5mM dithiothreitol at 56 °C for 30 minutes and alkylated using 11 mM iodoacetamide for 15 minutes at room temperature in darkness for absorption. The urea concentration in the protein samples were diluted to less than 2 M applying 100 mM triethylammonium bicarbonate. After the above treatments, trypsin was applied for the first digestion overnight at a trypsin to protein mass ratio (1: 50), and then for the second digestion for 4 hours at a trypsin to protein mass ratio (1: 100).
TMT labeling, HPLC fractionation, and LC-MS/MS analysis
The peptides were desalinated through the Strata X-C18 SPE column (Phenomenex) and vacuum dried, following digestion with trypsin. Peptides were reassembled into 0.5M triethylammonium bicarbonate and operated for the 10-PLEX TMT package according to the instructions of manufacture for the TMT/iTRAQ-kit. In short, one unit of the TMT/iTRAQ mixture was thawed and reassembled into 24μl acetonitrile (defined as the volume of mixture needed to mark of 100μg proteins). The peptide solutions were incubated for 2 hours at room temperature, pooled, desalted, and dried through vacuum centrifugation.
Using an Agilent-300 Extend C18 column (5 μm particles, 4.6 mm ID, 250 mm length), the samples were fractionated into various fractions through the high-pH reverse phase HPLC. In brief, peptides were initially separated in 10 mM ammonium-bicarbonate (pH-10) for 80 min into 80 fractions with gradient of 2% to 60% acetonitrile. Later, the peptides were combined into 9 fractions and dried by vacuum centrifugation.
The tryptic peptides were dissolved in the solvent-A (0.1 % formic-acid, 2 % acetonitrile), and straightly loaded into a home-made reversed phase analytical column (20 cm length, 100 μm i.d.). The gradient was comprised of an increasing from 6% to 22% solvent-B (0.1% formic acid in 90% acetonitrile) during 38 minutes, 22% to 32% in 14 minutes, and an increase to 80% for 4 minutes, then maintaining at 80% for the last 4 minutes. All processes were operated at stable flow rate of 450 nL/min using the EASY-nLC 1200 UPLC system.
The peptides were subjected to the NSI sources in Q-ExactiveTM HF X (Thermo), followed by the tandem-mass spectrometry (MS/MS) together online with the UPLC. The applied electrospray tension was 2.0 kV. The m/z assay size for a complete scan was 350 to 1600, and the Orbitrap detected the whole peptides at resolution of 120, 000. The peptides have been chosen for MS/MS with the NCE setting at 28. The fragments were identified at a resolution of 30,000 in the Orbitrap. A data dependent process that exchanged from one MS-scan to 20 MS/MS dynamic exclusion scans using 30.0 s. Automatic gain-control (AGC) was fixed at 1E5. The first set mass was fixed at 100 m/z.
Bioinformatics analysis of proteomic data
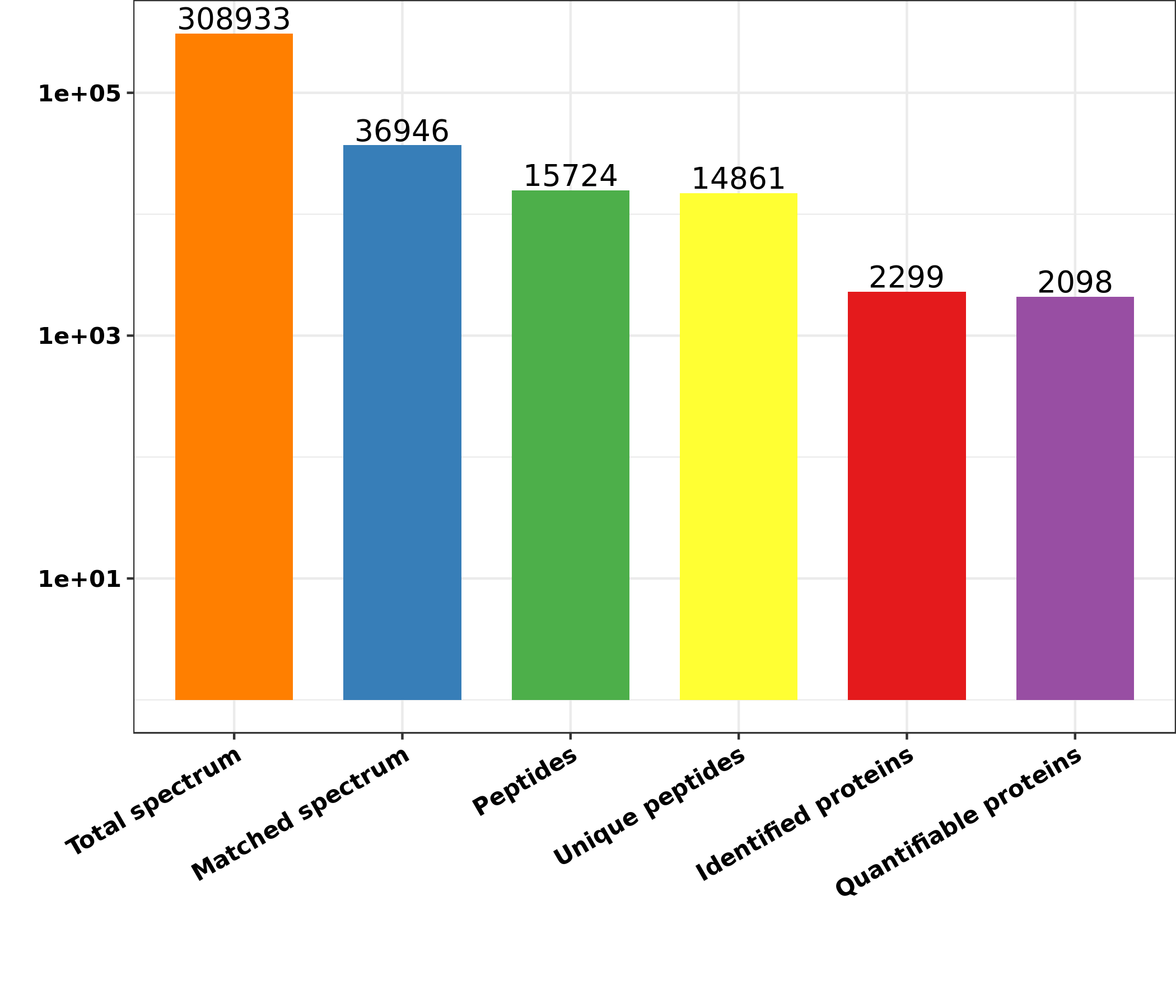
The MS/MS data was analyzed applying the explore engine Max-Quant (v-1.5.2.8). In Capra aegagrus hircus database concatenated using the reversed decoy database, the tandem mass spectra were detected. Trypsin/P has been determined as the cleavage enzyme which allows up to 2 lacking cleavages. First, the mass tolerance of precursor ions was fixed at 20 ppm in first check and 5 ppm was in second check. The mass tolerance for fragments ions were fixed at 0.02 Da. Carbamido-methyl on Cys was clarified for a set direction, and changeable directions were specified for oxidation on Met and acetylation on the protein N-term. FDR was changed to < 1 % and a lowest score was fixed to > 40 for the modified peptides. The minimum length of a peptide was fixed at 7. The TMT 10-PLEX was chosen for the quantification process. In MaxQuant, the parameters were fixed as the default values. The P values were calculated using the t-test of the two-sample two-tailed Student. Proteins with fold change of > 1.50 and P value <0.05 were identified as up-regulated DEPs between the high motility group and the low motility group. However, the proteins with fold change of <0.667 and P value < 0.05 were identified as the down-regulated proteins.
The Gene Ontology (GO) annotation was performed based on the UniProt GOA database (http://www.ebi.ac.uk/GOA/). At first, the IDs of identified proteins were transformed to UniProt-IDs, mapping to GO IDs with the protein IDs. Unless some identified proteins can be annotated by the UniProt-GOA database, the InterProScan will be applied to annotate protein’s GO functionality using the protein sequence alignment procedure. Based on the UniProt-GOA database, the DEPs were classified into three types: biological procedure, cell compartment, and molecular functions. For each type, two tailed Fisher’s exact-test was used to test the enrichment of the DEPs against all identified proteins. The GO with a modified P value < 0.05 was considered significantly. In addition, the information related to subcellular localization of the obtained DEPs was inferred with Wolfpsort (http:/www.genscript.com/psort/wolf psort.html).
The KEGG online service tools KAAS (http:/www.genome.jp/kaas-bin/kaas) was used to identify pathways that the obtained DEPs are enriched. Firstly, KAAS was applied to annotate the KEGG database description of the identified proteins. Then, the annotation results were mapped on the KEGG pathway database applying the KEGG online service tools KEGG mapping. Based on the KEGG database, the two tailed Fisher’s exact-test was used to detect the enriched channels to test the enrichment of DEPs against the entire detected proteins. The pathway was considered significantly with a corrected P value <0.05. According to the KEGG website, these pathways were specified into hierarchical groups.
The analysis of protein domain was conducted applying the InterPro domain database (http:/www.ebi.ac.uk / interpro/). For the identified proteins in each category, the InterPro (resources that allows functional evaluation of protein sequencing by identifying proteins in various groups and estimating the existence of domains and major locations) database have been scanned, and two tailed Fisher’s exact-test has been applied to analyze the enrichment of DEPs against those identified proteins. Proteins domain with a corrected P value < 0.05 was thought significant.
Parallel reaction monitoring (PRM) validation
The seminal plasma separation and total protein exaction were the same as the above procedure. The digested peptides were submitted to the PRM analysis. PRM is a newly developed approach to verify proteins using the quadrupole-Orbitrap mass spectrometer 38,80. In brief, the tryptic peptides were mixed in the solvent A and eluted in a reversed phase analytical column using the gradient solvent B (6 %-25 % over 40 minutes, 25 %-35 % over 12 minutes, 80 % over 4 minutes, and 80 % over the last 4 minutes) at a rate of 500 nL/min. The peptides measured (1.5 mg per sample) were analyzed with an online Q ExactiveTM plus the Orbitrap mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA) coupled with the UPLC. The applied electrospray tension was 2.2 kV. The full MS scans (400-960 m/z) were obtained at a resolution of 70,000 using AGC of 3E6 and a highest injection time (MIT) of 50 ms. A data independent protocol (one MS scan followed by 20 MS/MS scans) was used for the MS/MS scans with the following parameters,: resolution, 17,500; NEC, 27; AGC, 1E5; MIT, 120 ms; insulation window, 1.6 m/z. The PRM data was analyzed using the Skyline 3.6. The results were quantified for every peptide, and the DEPs detected were screened and compared with the MS data derived from the TMT.
Seminal plasma metabolite exaction
In brief, the collected seminal plasma samples were first warmed on ice. The samples were vortexed for 30 seconds to ensure complete mixing. Then, 3 volumes of ice-cold methanol were added to 1 volume of seminal plasma, followed by vortexing for 3 minutes. The mixture was further centrifuged at 12,000 g for 10 minutes at 4 oC. The supernatant was collected and centrifuged at 12,000 g for 5 minutes at 4 oC again. After filtration through a 0.22 µm filter membrane, the supernatants were transferred into the injection bottles. Finally, the samples were preserved at -80 oC prior to the LC-MS/MS analysis. In addition, the pooled QC samples were simultaneously prepared by mixing 10 μL of each exacted mixture.
HPLC
The exacted seminal plasma samples were analyzed using the LC-ESI-MS/MS system (UPLC, Shim-pack UFLC SHIMADZU CBM A system, https://www.shimadzu.com/; MS, QTRAP 6500+ System, https://sciex. com/). The analytical parameters were set as following: UPLC: column, Waters ACQUITY UPLC HSS T3 C18 (1.8 µm, 2.1 mm×100 mm); the column temperature, 35 oC; the flow rate, 0.3 mL/min; the injection volume, 1 μL; the solvent system, water (0.01 % methanolic acid): acetonitrile; the gradient program of positive ion, 95:5 (V/V) at 0 minutes, 79:21 (V/V) at 3.0 minutes, 50:50 (V/V) at 5.0 minutes, 30:70 (V/V) at 9.0 minutes, 5:95 (V/V) at 10.0 minutes, and 95:5 (V/V) at 14.0 minutes; the gradient program of negative ion, 95:5 (V/V) at 0 minutes, 79:21 (V/V) at 3.0 minutes, 50:50 (V/V) at 5.0 minutes, 30:70 (V/V) at 9.0 minutes, 5:95 (V/V) at 10.0 minutes, and 95:5 (V/V) at 14.0 minutes.
ESI-QTRAP-MS/MS
The LIT and triple quadrupole scans were acquired on a triple quadrupole-linear ion trap mass spectrometer (QTRAP) (QTRAP 6500+ LC-MS/MS System) installed with an ESI Turbo Ion-Spray interface and controlled by the Analyst 1.6.3 software (Sciex). The QTRAP was operated in both positive and negative ion modes. The parameters of the ESI source operation were set as the following: the source temperature: 500 oC; the ion spray voltage (IS): 5500 V (positive) and -4500 V (negative); the ion source gas I (GSI): 55.0 psi; the gas II (GSII): 60.0 psi; the curtain gas (CUR): 25.0 psi; the collision gas (CAD): high. The instrument tuning and mass calibration were performed in 10 and 100 μmol/L polypropylene glycol solutions using the QQQ and LIT modes, respectively. A specific set of MRM transitions were monitored during each period based on the metabolites eluted within this period.
Metabolomics data analysis
The original data files acquired by the LC-MS analysis were first converted into mzML format using the ProteoWizard software. Peak extraction, alignment, and retention time correction were performed by the XCMS program. The “SVR” method was used to correct the peak area. The peaks were filtered in accordance with a deletion rate > 50% in each group of samples. After the above treatments, the identified metabolite information was obtained by searching the laboratory’s self-built database, the public database (Metlin), and metDNA. Finally, the statistical analysis was carried out by the R program. The statistical analysis includes the univariate analysis and the multivariate analysis. The univariate statistical analysis was performed using Student’s t-test and variance multiple analysis. The multivariate statistical analysis was carried out using these approaches including principal component analysis (PCA), partial least squares discriminant analysis (PLS-DA), and orthogonal partial least squares discriminant analysis (OPLS-DA).
Statistical analysis
The data associated with motility, plasma membrane and acrosome integrity of goat sperm were statistically analyzed by T-test using the JMP10.0 software (SAS Institute Inc., Cory, NC, USA). Data normality and homogeneity of variances were verified using the Shapiro–Wilk normality tests and Levene’s tests, respectively. The data were presented as means ± SEM. It was thought that the data with value of P<0.05 or P<0.01 was statistically significant.
{kind=link}