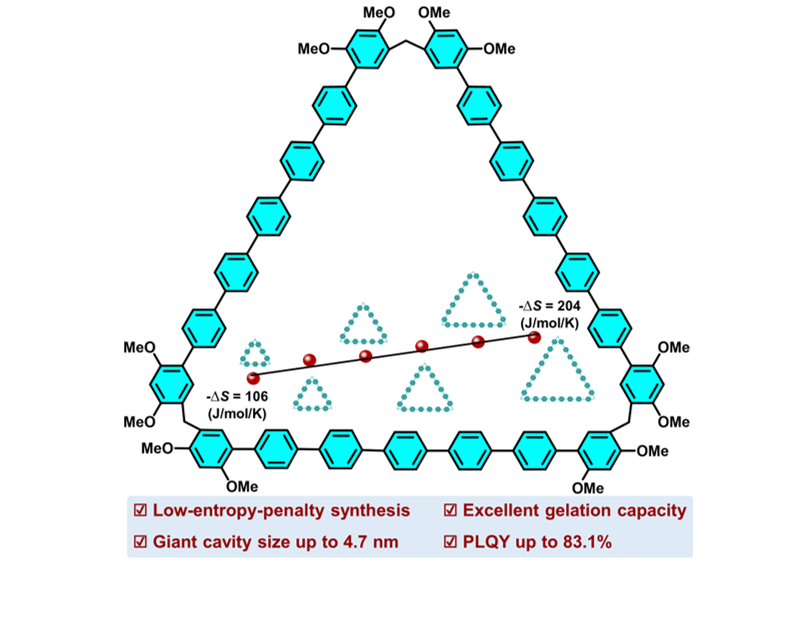
We first calculated the entropy penalty (-ΔS)53 resulting from extending the monomer length of p-oligophen[n]arenes (our strategy), and compared it to that of increasing monomer numbers of pillar[n]arenes and calix[n]arenes (conventional strategy) (Figs. 1 and S2). From pillar[5]arene to pillar[10]arene, the number of phenyl units in macrocycle increased from 5 to 10, the -ΔS increased 7.9-fold, and the cyclization yields reduced from 71–2% (Fig. 1a).44,48 Conversely, from terphen[3]arene to octaphen[3]arene, the number of phenyl units in macrocycle increased from 9 to 24, and the -ΔS only increased 0.9-fold, indicating the great advantages of the proposed strategy (Fig. 1b).
We then verify the effectiveness of our strategy experimentally. By coupling 4,4''-dibromoterphenyl with two 2,4-dimethoxyphenylboronic acid reaction modules, a rigid quinquephenyl monomer (M1) was synthesized in 81% yield (Scheme S2, and Figures S3-5). Monomer M1 was further condensed with paraformaldehyde ((CH2O)n) using a Lewis acid catalyst to form cyclic products quinquephen[3–6]arenes (QP[3–6]). This cyclization reaction can be performed in various solvents, such as 1,2-dichloroethane (DCE), tetrachloroethane (TCE), dichloromethane (DCM), and CHCl3, and catalysts, such as BF3·Et2O, FeCl3, AlCl3, and trifluoromethanesulfonic acid (TfOH)) (Table S1). Under optimized conditions of using DCE as the solvent and BF3·Et2O as the catalyst (8%), and at 25 °C for 2 h, QP[3–6] can be obtained with an overall yield of 72% (44% for QP[3], 23% for QP[4], 5% for QP[5], and trace amounts for QP[6]) (Schemes 1a, S1, S3, and Figures S6–15). This relatively high cyclization yield indicates that our strategy is significant in producing giant macrocycles.54 We further expanded the monomer size and synthesized a dihexylheptaphenyl monomer (M2) (Schemes S4–6, and Figures S16–24). It should be noted that two n-hexyl groups were introduced to the monomer to increase its solubility. Utilizing similar cyclization conditions, heptaphen[3–6]arenes (HP[3–6]) were synthesized in 45% yield for HP[3]; trace amounts for HP[4–6] were detected using high-resolution mass spectrometry (HR-MS) (Schemes 1b, S7, and Figures S25–30). Although the cyclic trimer is the dominant product, HP[3] has a skeleton consisting of 21 phenyls, much larger than traditional macrocyclic arenes.
Single-crystal structures provided clear insights into the conformations and atomic-level structures (Scheme 1, and Table S2). QP[3] exhibited a side length of 2.0 nm, and the macrocycle had a relatively electronegative cavity (Figure S31). However, we did not obtain single crystals of QP[4–6] and HP[3–6]; therefore, the calculated structures were obtained using the Mopac2016 program method. From QP[4] to QP[5] and QP[6], the macrocyclic sizes increased from 2.0 nm to 3.2 nm and 3.8 nm, respectively (Figures S32–34). In the case of larger macrocycles HP[3–6], their macrocyclic sizes increased from 2.8 nm for HP[3] to 3.3 nm for HP[4], 4.6 nm for HP[5], and 4.7 nm for HP[6] (Scheme 1b, and Figures S35–38). Giant cavity sizes demonstrate the advantages of the proposed strategy. Furthermore, the surface electrostatic potential maps proved that all the macrocycles possessed a relatively electronegative cavity (Figures S31b–38b).
In addition to the 2,4-dimethoxyphenyl reaction module, the 2,5-dimethoxyphenyl reaction module was evaluated. Since linear monomer cannot give any cyclic products, we then used a V-shaped 2,5-dimethoxyquinquephenyl monomer (M3) with an angle of 120°. It produced a hexagonal macrocycle (2,5-QP[3], 17% yield) and a saddle-shaped macrocycle (2,5-QP[4], 5% yield) (Schemes 2a, S8–12 and Figures S39–51). Single-crystal structures confirmed that even the cyclic trimer (2,5-QP[3]) possessed a giant cavity size of 2.0 nm, proved the good universality of our strategy for constructing giant macrocycles (Scheme 2a).
By constructing 2,5-dimethoxyheptaphenyl monomers (M4 and M5), the monomer sizes could be expanded and larger giant macrocycles of 2,5-dimethoxyheptaphen[n]arenes (CN-HP[3–5] and CHO-HP[3–4]) could be produced under similar conditions (Schemes 2b, S13–18, and Figures S52–73). For CN-HP[3–5], the cyclic trimer CN-HP[3], tetramer CN-HP[4], and pentamer CN-HP[5] were obtained in 15%, 8%, and 4% yields, respectively (Scheme S16, and Figures S59–64). For CHO-HP[3–4], the cyclic trimer CHO-HP[3] and tetramer CHO-HP[4] were obtained in yields of 12%, and 6%, respectively (Scheme S18, and Figures S68–73). Furthermore, all the macrocycles possessed relatively electronegative cavities (Figures S74–78). Modifiability is crucial for the further development of host–guest recognition, assembly, and functional applications; the preset CN and CHO groups in these macrocycles make it convenient to construct assembly and functional materials. We also examined the feasibility of modifying the giant macrocycles through post-modification. As a typical example, BBr3 was gradually added to a solution of 2,5-QP[3] in DCM and stirred at 0 °C for 3 days under a nitrogen atmosphere. The per-hydroxylated macrocycle 2,5-QP[3]-OH was obtained in 97% yield after quenching and drying (Scheme S19 and Figures S79–81). This result verified the good modifiability of the synthesized giant macrocycles because further modification could be easily performed via reactive OH groups.
During the purification of QP[3] and cultivation of its single-crystal structure, a supramolecular gel was formed in DCM/hexane and THF/hexane. This intriguing phenomenon suggests that QP[3] probably has a good assembly capacity. Under typical conditions, 0.5 mL of DCM solution dissolving 5 mg of QP[3] was dropped 0.4 mL of hexane, resulting in the immediate formation of a gel. Conversely, monomer M1 was unable to form such a gel (Figure S82). HP[3] also formed an organogel under similar conditions (1:1 DCM/hexane) (Figs. 2c–d, and g–h). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used for morphology characterization. Both xerogels of QP[3] and HP[3] were fibrous with length and width of 10–100 µm and 20–300 nm, respectively (Figs. 2a–d). Monomers M1 and M2 exhibited disordered structures (Figs. 2e–h). Powder X-ray diffraction (PXRD) experiments demonstrated the non-crystalline state of the xerogels, and a broad peak at approximately 2θ = 20° indicated the existence of π-π interactions (Figure S83).55
To elucidate the assembly mechanism, we evaluated two additional cyclic trimers: quaterphen[3]arenes (QT[3]) and terphen[3]arenes (TP[3]). The monomer units of QT[3] and TP[3] possessed one and two fewer phenyls than that of QP[3], respectively (Figs. 2j). Under the same gelation conditions, TP[3] remained a clear solution, QT[3] produced some precipitates, and QP[3] formed a non-flowing white gel even when the bottle was inverted. This indicated that with an increased in the macrocycle size, the assembly capacity increased. The single-crystal structures of TP[3], QT[3], and QP[3] provided more information about the assembly at the atomic level (Figures S84–87, and Table S4). In the solid state, every QP[3] molecule (cyan) was tightly stacked with the other five molecules (red, purple, orange, and gray) via thirteen phenyls and provided multiple π-π interactions sites (2.7, 2.8, 2.9, 3.1, 3.1, 3.1, 3.1, 3.2, 3.2, 3.2, 3.3, 3.4, 3.6, and 3.8 Å) (Fig. 2k). One edge of the triangular QP[3] (cyan) completely overlapped with that of another (orange). Conversely, the remaining two edges were stacked with two other parallel molecules (red and purple) and complemented by additional molecules (gray) (Figures S84a and S85). For smaller macrocycle QT[3], every molecule (orange) was stacked with three other molecules (gray) via six phenyls and provided limited number of π–π interactions sites (3.1, 3.1, 3.5, 3.5, 3.9, and 3.9 Å) (Figures S84b, and 86). The smallest molecule TP[3] interacts with other molecules (gray) via only four phenyls and provides the least number of π–π interactions sites (2.9, 3.2, 3.2, 3.8, and 3.8 Å) (Figures S84c, and S87). These results clearly revealed that larger macrocycles could provide more π–π interaction sites and therefore better assembly capacity. Accordingly, the largest macrocycle QP[3] facilely formed organogel, moderate macrocycle QT[3] produced precipitates, and the smallest macrocycle TP[3] remained a clear solution.
These macrocycles also exhibit intriguingly photophysical properties. QP[3] had a strong blue emission peak at 408 nm and a slight red shift compared to the monomer M1 in the DCM solution (Figs. 3a and S88a). The UV-Vis spectra showed similar peaks at 316 nm for QP[3] and 319 nm for M1 (Figure S88b). These results proved that the methylene bridging had a limited effect on the ground state but a significant effect on the excited states of the quinquephenyl units. After assembly into the xerogel, the emission of QP[3] exhibited a red shift to 414 nm, whereas monomer M1 in the solid state had an emission identical to that in the solution (400 nm) (Figs. 3a and S88c). This result indicates that the assembly of the xerogel is a J-aggregate which was consistent with the packing mode of single-crystal structures.56,57 The delay-time photoluminescence spectra showed a dual emission with peaks at 414 nm and 508 nm. As the delay time increased from 0.2 ms to 5 ms, the peak at 414 nm sharply disappeared and the peak at 508 nm remained. This observation hints a phosphorescent nature of the QP[3] xerogel, further proved by the millisecond lifetime (0.35 ms) (Figs. 3b and S88d). Time-resolved PL decay spectra proved the fluorescence lifetimes of QP[3] (peak at 414 nm, τ = 1.1 ns) and M1 (peak at 400 nm, τ = 0.86 ns) (Figure S89a-b). HP[3] xerogel exhibited similar emission with a dual emission of intense fluorescence (peak at 388 nm, τ = 1.1 ns) and phosphorescence (peak at 534 nm, τ = 0.37 ms), which was further supported by the delay-time photoluminescence spectra (Figs. 3b, S89d and S90a). In a DCM solution, HP[3] exhibited an absorption peak similar to that of monomer M2 and an intense blue emission (Figure S90b–c). Notably, the photoluminescence efficiencies were 72.8% for QP[3] and 83.1% for HP[3] in the xerogels, far higher than those of their monomers (43.1% for M1 and 33.5% for M2) (Figs. 3c and S91–92).
This remarkable emission enhancement inspired us to determine the origin. According to Kasha’s rule, an excited molecule generally decays fast and highly efficiently to the lowest excited singlet S1. Subsequently, S1 undergoes three competing decay processes: radiative decay to the S0 by emitting fluorescence with a rate constant \({\text{k}}_{r}^{F}\), decay to the ground state S0 via nonradiative process with a rate constant \({\text{k}}_{nr}^{F}\), and conversion to the triplet state (Tn, n ≥ 1) with an intersystem crossing rate constant k𝑖𝑠𝑐. T1 further decays to S0 via a radiative process of phosphorescence with a rate constant \({\text{k}}_{r}^{P}\)or a nonradiative process with a rate constant of \({\text{k}}_{nr}^{P}\).58 For these giant macrocycles, the radiative decay rate constant of the singlet state was enhanced to \({\text{k}}_{r}^{F}\) = 6.7 × 108 s−1 for macrocyclic xerogels QP[3], which is larger than that of the monomers powder of M1 (\({\text{k}}_{r}^{F}\) = 5.0 × 108 s−1) (Table S5). Such improvements were also found in macrocyclic xerogels HP[3] ( \({\text{k}}_{r}^{F}\) = 7.6 × 108 s− 1 ) and M2 ( \({\text{k}}_{r}^{F}\) = 1.5 × 108 s−1 ). Moreover, the nonradiative decay rate constants of the macrocyclic xerogels (\({\text{k}}_{nr}^{F}\)= 2.5 × 108 s−1 for QP[3], and \({\text{k}}_{nr}^{F}\)= 1.5 × 108 s−1 for HP[3]) smaller than that of the monomers powder (\({\text{k}}_{nr}^{F}\)= 6.6 × 108 s−1 for M1, \({\text{k}}_{nr}^{F}\)= 2.9 × 108 s− 1 for M2) (Table S5). The larger \({\text{k}}_{r}^{F}\) and smaller \({\text{k}}_{nr}^{F}\) of the macrocyclic xerogels indicated that the macrocyclic assembly significantly suppressed the nonradiative decay and promoted the radiative decay of QP[3] and HP[3]. Because τp = 1/(\({k}_{r}^{P}\)+ \({k}_{nr}^{P}\)), the similar phosphorescence lifetimes between macrocyclic xerogels and monomers powder indicated that their \({k}_{r}^{P}\)+ \({k}_{nr}^{P}\) were near identical in a disordered monomer and macrocyclic assembly. This reveals that the assembly has negligible effects on their triplet state radiative and nonradiative decay processes. Based on these photophysical properties and aforementioned assembly investigation, a possible mechanism for the emission enhancement of macrocyclic assembly was proposed. Macrocycles QP[3] and HP[3] were strictly confined in the xerogels assembly via multiple intermolecular π-π interactions. Accordingly, their nonradiative decay processes of rotation/vibration were restricted, the radiative decay process of fluorescence was boosted, and eventually their photoluminescent quantum efficiencies were promoted (Figs. 3d).
{kind=link}