Protein-protein interactions (PPIs) are at the core of all cellular processes and have the capacity to infer functions of unknown proteins. PPI analyses are done through a plethora of methods, much of which are performed in heterologous hosts or in vitro1. By contrast, the assessment of PPIs in native hosts is largely restricted to different affinity-based purification schemes or what is referred to as proximity labelling. Proximity labelling may be done by fusing a protein of interest, referred to as bait, with an enzyme that can label proteins in the vicinity of the bait with a certain substrate. The efficiency of this process depends on the expression level of the enzyme-tagged bait and the substrate, and on favorable conditions to catalyze the labelling. The most frequently used proximity labelling entails various versions of biotin ligases or peroxidases, including for example TurboID2, 3. Some of the drawbacks of these systems include the need to add the substrate (e.g. biotin) externally, timing and temperature optimization to activate the substrate and the ability to control the labeling reaction. In addition, endogenous biotin and peroxidases may confound results in plant cells2, 3. However, recent developments have produced a completely genetically encoded proximity labelling system using pupylation, called PUP-IT (Fig. 1a)4.
The PUP-IT system is based on the prokaryotic enzyme PafA, which activates, holds and catalyzes the attachment of a PUP(E) peptide to lysine residues of proteins in close vicinity of the PafA4. Pupylation is absent in eukaryotes, and thus PUP-IT may be used to label proteins without labelling background4. In plant science, PUP-IT has recently been used in transient protoplast assays and in stable Arabidopsis lines with only one bait5, 6. Here, we tested the PUP-IT system on a broader scale by using cytosolic and integral membrane bait proteins in a range of plant systems, including Nicotiana benthamina transient assays, Arabidopsis PSB-D suspension cells and Arabidopsis thaliana stable transgenic lines. We also added different tags on the Pup(E), e.g. FLAG-Pup(E) and StrepII-FLAG-Pup(E) providing several strategies for affinity-based protein purification of Pup(E)-tagged proteins. Finally, we provide a versatile cloning strategy with inducible- or ubiquitous promoters driving PafA and Pup(E) on the same vector backbone, which allows implementation of the system via a single transformation event.
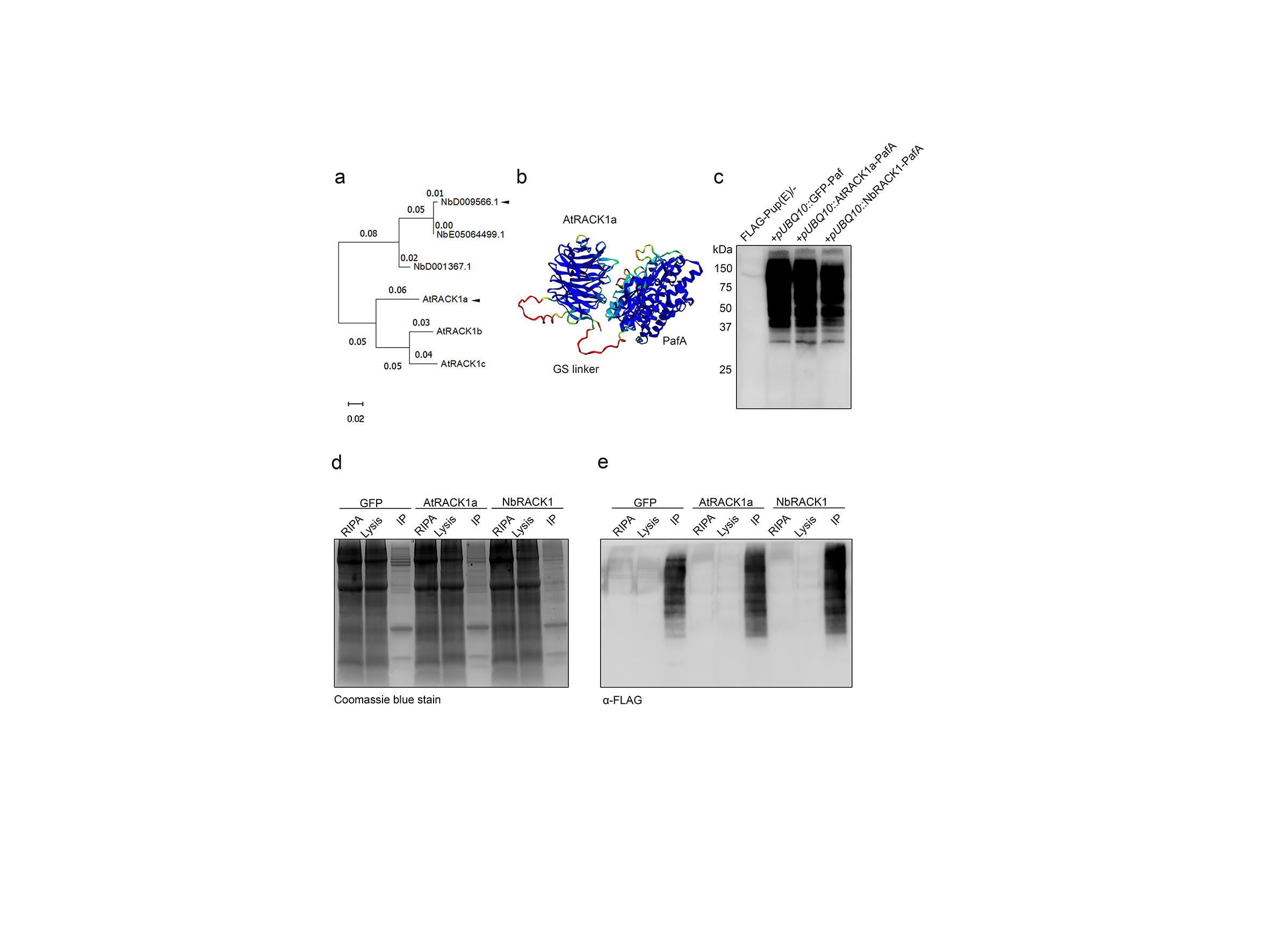
We first tested the PUP-IT system using the scaffold protein RECEPTOR FOR ACTIVATED C KINASE1 (RACK1) as bait in transient expression assays in N. benthamiana leaves. RACK1 has a plethora of interactors7, mainly associated with protein translation and ribosome functions. RACK1 has three paralogs in Arabidopsis, of which RACK1A (AtRACK1a) is the most studied7. We therefore selected AtRACK1a and one of the N. benthamiana homologs (Fig. S1a), referred to as NbRACK1, as baits fused to PafA in our PUP-IT approach, with GFP fused to PafA as control (Fig. 1b). We introduced a flexible linker between the PafA and the RACK1 to minimize interference of the PafA on the native bait function. We based the linker length and content (glycine-serine linkers) on the Alphafold2 predicted structure of PafA and AtRACK1 to ensure flexibility and thus functionality of the linked proteins (Fig. S1b)8, 9, 10, 11. We infiltrated the agrobacteria carrying the constructs into N. benthamiana leaves, performed subsequent enrichment of FLAG-PUP-labelled proteins and undertook mass-spectrometry (MS) analyses to identify target proteins of the different RACK1s (Fig. S1c-e). We next screened for proteins detected exclusively, or highly enriched, in the RACK1-PafA samples compared to GFP-PafA, and calculated fold enrichment and statistics to identify proteins that potentially interacted with RACK1-PafA. From this approach, we found 254 putative interactors of the AtRACK1a and 259 of the NbRACK1 compared to the GFP control (Fig. 1c) (Table S1). Importantly, we found that 154 of these proteins overlapped between the AtRACK1a and NbRACK1 samples (Fig. 1c), corroborating our expectation that the RACK1s in Arabidopsis and N. benthamiana engage with similar proteomes. To assess what processes were enriched among these proteins we next undertook Gene Ontology (GO) analyses and found that terms related to protein translation, RNA binding and ribosome function were among the most enriched for the proteins in line with the known activity of RACK1 (Fig. 1d). To further corroborate these inferences, we looked at overlap between the PUP-IT identified interactors and known interactors. To do this, we identified Arabidopsis homologs of the potential RACK1 interactors from the N. benthamiana PUP-IT experiment using BLAST and selected the best score hits. We then used these homologs to search the RACK1 interactome via the STRING database (https://string-db.org/). Out of the 154 proteins enriched from our RACK1 experiments, we found 51 proteins closely connected to RACK1 in STRING (Fig. 1e, network type: physical subnetwork, active interaction sources: experiments). In addition, several other interacting proteins formed smaller connected satellite units. It is important to note that although the BLAST analyses may have identified true orthologs between Arabidopsis and N. benthamiana, it is also possible that some proteins are part of more complex protein families, which may lead to an underestimation of positive hits.
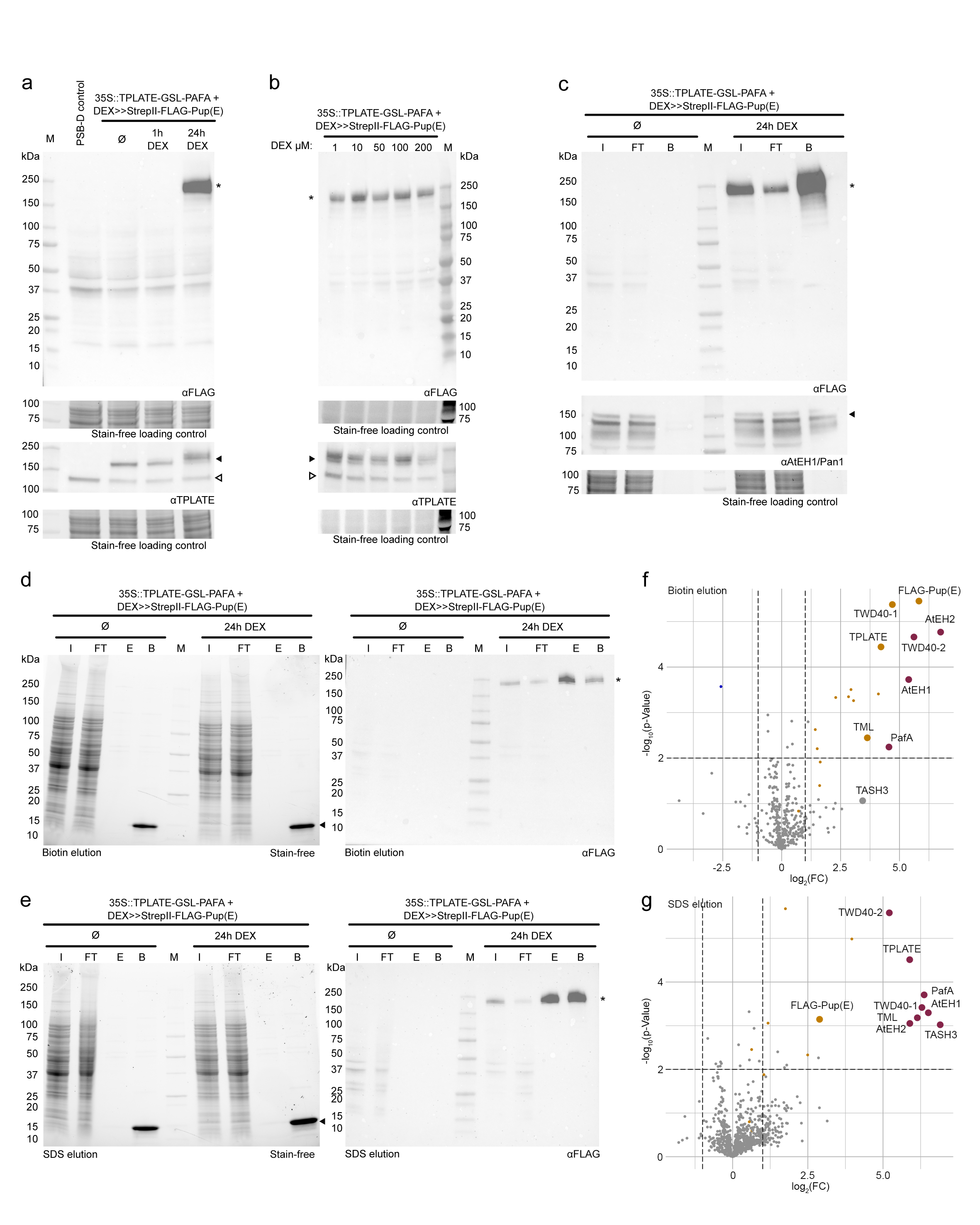
We next tested the PUP-IT approach in another commonly used system for proteomics in plant biology; Arabidopsis PSB-D suspension cells. Here, we chose to investigate the TPLATE complex (TPC), which is a key endocytic protein complex in plant cells containing eight subunits12. We, therefore, fused the PafA with TPLATE via a GSL linker and combined it with the expression of StrepII-FLAG-Pup(E) under a dexamethasone (DEX)-inducible promoter on the same T-DNA (Fig. 1f). We first optimized the time points and concentrations of DEX treatment by detecting the StrepII-FLAG-Pup(E) labelled TPLATE bait (Fig. S2a-b). Next, we affinity-purified the pupylated proteins and probed the bead fractions with an antibody against AtEH1/Pan1, a subunit of the TPC. The co-purification of the AtEH1/Pan1 subunit served as a first proxy for complex incorporation of the tagged bait (Fig. S2c). We also tested whether we could elute the majority of StrepII-FLAG-Pup(E) labelled proteins with 50 mM biotin while minimizing Streptactin contamination, which could interfere with mass spectrometry analysis (Fig. S2d). In parallel with the biotin elution strategy, we also used an SDS elution procedure (Fig S2e). Subsequent proteomic analyses showed that PUP-IT has the capacity to specifically isolate the TPLATE complex as various subunits of the TPLATE complex were significantly enriched or even exclusively found in the DEX group (Fig. S2f-g) (Table S2). Furthermore, the absence of the FLAG-PUP(E) peptides in the control group indicates that the DEX-inducible system in PSB-D is not leaky and therefore allowed tight control over the reaction (Fig. S2f-g). Affinity Purification-Mass Spectrometry (AP-MS) and TurboID-based biotin-proximity labelling have previously been performed on TPLATE in the same system, which allowed us to compare PUP-IT to these approaches12, 13. To do this, we undertook intensity-based absolute quantification (iBAQ)14 of TPLATE subunits normalized to TPLATE as measure of enrichment of the subunits for each of the different experiments. We found that the subunits were substantially more enriched in the PUP-IT-based approaches compared to both AP-MS and the TurboID assays, highlighting high specificity of PUP-IT in labelling known TPLATE interactors (Fig 1g; Table S2).
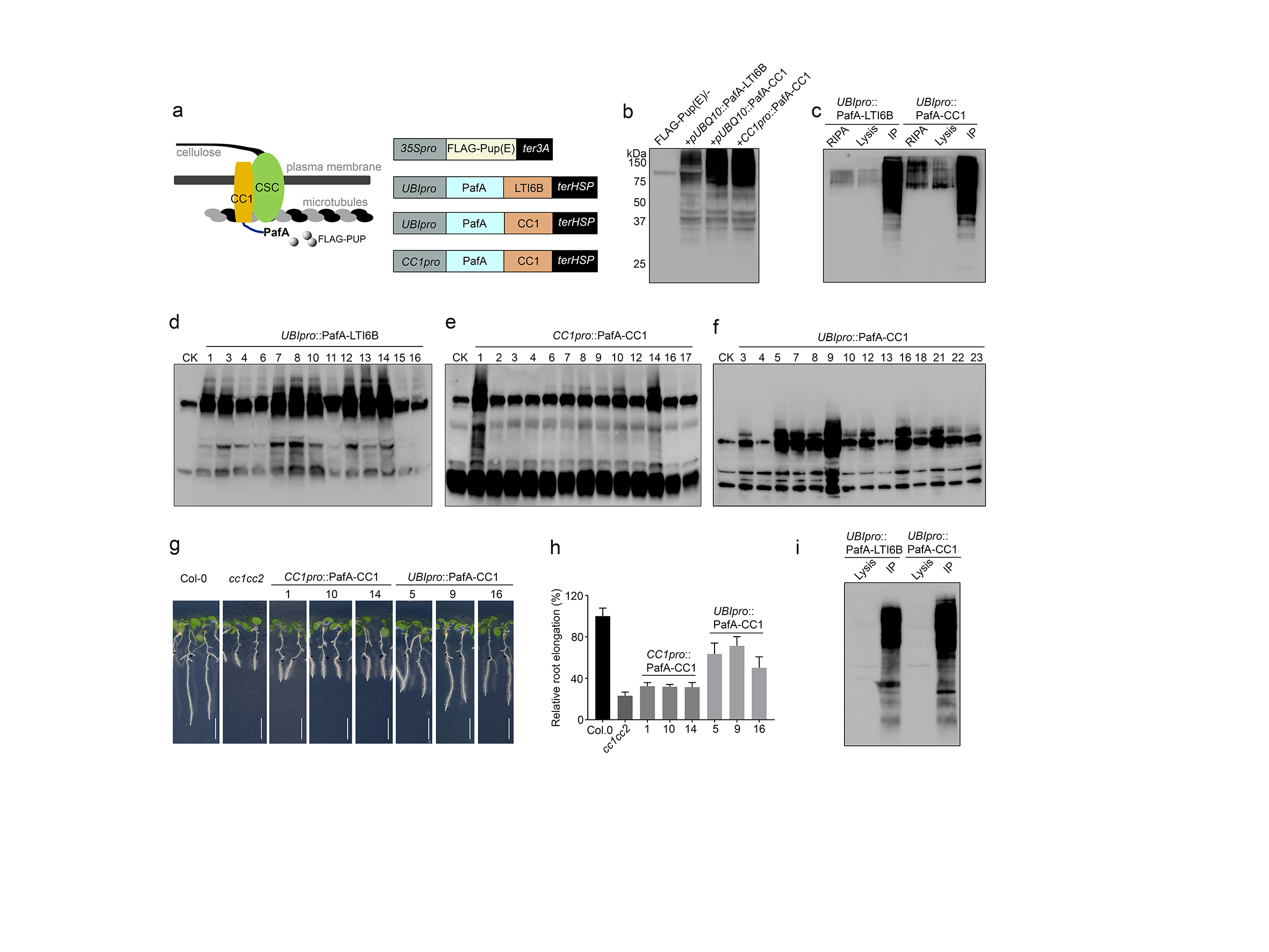
The above examples demonstrate the utility of PUP-IT as an effective proximity labelling system in plant biology. We next employed the PUP-IT system to investigate the membrane-based CELLULOSE SYNTHASE (CESA) complex (CSC), which underpins cellulose synthesis in plant cell wall biology15. CSC has many known interactors; however, mechanisms that regulate CESA activity and trafficking are still unclear, suggesting that we are missing important interactors. Here, we chose to fuse the PafA to the N-terminus of COMPANION OF CESA 1 (CC1; Fig. S3a), which is one of the central components of the CSC16 and transformed the construct either into N. benthamiana leaf cells (transient infiltration) or into cc1cc2 double mutant Arabidopsis plants (stable transformation). As the CSC is largely present at the plasma membrane, we used the plasma membrane-localized protein LOW TEMPERATURE INDUCED PROTEIN 6B (LTI6B) as control (Fig. S3a). We first analyzed the enriched proteins from the N. benthamiana infiltration and found that many of the known CESA complex proteins were enriched in the CC1 samples as compared to the LTI6B (Fig. 2a, Fig. S3b-c) (Table S3). For example, beyond CC1, we found N. benthamiana proteins corresponding to the main CESAs that make up the core of the complex (e.g. CESA1), as well as CESA INTERACTING1 (CSI1) that connects the CSC to underlying microtubules17. In addition, we found SHOU4-LIKE that regulates trafficking of the CSC, as well as the STRUBBELIG-RECEPTOR FAMILY6 (SRF6) that is involved in response to cellulose deficiency (Fig. 2a) (Table S3)18, 19.
With these promising results, we next generated Arabidopsis stable transgenic lines usingeither a CC1 promoter- or Ubiquitin10 promoter (UBI10pro)-driven PafA-CC1 construct to transform cc1cc2 double mutant plants. We used plants expressing a UBI10pro-driven PafA-LTI6B construct as control. We screened independent transgenic lines to obtain suitable levels of PafA-LTI6B and PafA-CC1 activity, and then selected lines based on the ability of the PafA-CC1 to complement the cc1cc2 mutant phenotype (Fig. S3 d-f). Here, we scored the phenotype based on growth on media plates supplemented with the cellulose synthesis inhibitor isoxaben, which leads to severe growth retardation of cc1cc2 mutant roots16. We found that while the CC1 promoter-driven constructs partially complemented the isoxaben-related phenotypes of cc1cc2, the UBI10pro-driven constructs complemented the phenotypes better (Fig. S3 g-h). Based on these results, and since we used the UBI10pro-driven LTI6b expression as control, we chose to use the UBI10pro-driven PafA-CC1 expression for our PUP-IT experiments. To ensure that we both had sufficient material for enrichment assays, as well as active cellulose synthesis, we used six-day-old seedlings as material for the FLAG-PUP enrichment (Fig. S3i). Similar to the transient infiltration assays in N. benthamiana leaves, we again found the components of the core cellulose synthesis machinery including CESAs (1, 3 and 6) and CSI (1 and 3)20, as well the SHOU4, SRF1 corresponding to their homologs SHOU4L and SRF6 identified in the N. benthamiana samples (Fig. 2a-b; Table S4). In addition, we found the subunits of TPLATE complex, which mediates CSC endocytosis, as well as the v-ATPase subunit DE-ETIOLATED3 (DET3) involved in CSC secretion and recycling at the trans-Golgi network (TGN)21, 22. We note that despite using different organisms, conditions, transformation approaches and organs, we identified the key core proteins of the CSC using the PafA-CC1 in N. benthamiana and Arabidopsis. However, the CC1 interactomes differ across the comparison, possibly indicating that the interactomes of a given protein vary depending on development, environment and biological system.
While some of the components of the CSC have been identified based on forward genetic screens, many of the more recently identified components have been found via co-expression analyses23. To see if any of the genes that encode identified proteins from the CC1 PUP-IT analyses were co-expressed with the CESA genes, we inspected their co-expression relationships. Here, we identified BFA-VISUALIZED ENDOCYTIC TRAFFICKING DEFECTIVE1 (BEN1)/ BREFELDIN A-INHIBITED GUANINE NUCLEOTIDE-EXCHANGE PROTEIN 5 (BIG5)/ Arabidopsis thaliana HopM interactor 7 (AtMIN7) as a promising candidate (Fig. S4a) (Table S3-4). In addition, several proteins previously reported to interact with BEN1, i.e. HYPERSENSITIVE TO LATRUNCULIN B1 (HLB1), Brefeldin A-inhibited guanine nucleotide-exchange protein 2 (BIG2) and Arabidopsis thaliana HopM interactor 10 (AtMIN10)24, were also enriched in the Arabidopsis PafA-CC1 samples as compared to PafA-LTI6B (Fig. 2b). We therefore aimed to place BEN1 function in context of the CC1 and the CSC.
BEN1 is an ADP-ribosylation factor (ARF) guanine nucleotide exchange factor (ARF-GEF) involved in the trafficking of proteins between the TGN/Early Endosomes (EE) and the plasma membrane25. To put the BEN1 localization in context to that of CC1, we co-expressed GFP-CC1 and BEN1-mCherry in stable Arabidopsis lines and observed that the fluorescent signals co-localized at the plasma membrane and in endomembrane compartments (Fig. 2c-d), in line with the function of both BEN1 and CC1 at the TGN25, 26. These results indicated that the proteins may function at the same locations inside the cell. To confirm interactions between the two proteins, we first co-expressed BEN1-mCherry with the FLAG-Pup(E) and PafA-CC1 in N. benthamiana. These experiments showed that PafA-CC1 also could pupylate BEN1 in this transient infiltration system (Fig. 2e). To further corroborate interaction between BEN1 and CC1, we performed co-immunoprecipitation (co-IP) assays using N. benthamiana leaves. Here, we found that a GFP-CC1 could co-IP BEN1-mCherry as compared to GFP (Fig. 2f). To rule out that the BEN1-mCherry enrichment was due to the interaction to CC1 and not to spurious GFP-mCherry interactions, we show that neither expression of GFP nor GFP-CC1 with mCherry revealed any co-IPed protein bands in the size region of BEN1-mCherry (Fig. S4b). Interestingly, the molecular weight of the enriched BEN1-mCherry protein was substantially larger in the output of the co-IP experiment (Fig. 2f). One reason for this might be that the enriched BEN1-mCherry is poly-ubiquitinated, which has been reported for BEN1 in context of other PPIs27. Poly-ubiquitinated proteins may be targeted for degradation by the proteasome, which may be inhibited by applying the molecule MG13228. To assess whether the enriched BEN1-mCherry was poly-ubiquitinated, we therefore treated the samples with the proteasome inhibitor MG132 and probed the co-IPed BEN1-mCherry with an antibody that recognizes poly-Ubiquitin, with positive signal (Fig. S4c). In addition and consistent with the notion that the poly-ubiquitinated BEN1-mCherry may be degraded, we also found that the treatment of MG132 led to an increased abundance of the poly-ubiquitinated BEN1-mCherry (Fig. S4d).
We next further confirmed the interaction between BEN1 and CC1 using split-ubiquitin yeast two hybird (Y2H) and N. benthamiana-based Bi-molecular Fluorescence Complementation (BiFC) assays. In the Y2H, we found that both CC1 and its C terminal truncation (fused to NubG) interacted weakly, but consistently, with BEN1 (fused to Cub). However, when we removed also the N-terminal region, we observed no interactions between the CC1 and BEN1 (Fig. 2g-h, and Fig. S4e). In the BiFC assays, we observed strong fluorescent signals when co-expressing nYFP-CC1 or nYFP-CC1∆C with BEN1-cYFP, but detected only faint fluorescence when co-expressing nYFP-CC1∆N and BEN1-cYFP (Fig. 2i). We quantified the fluorescence from the BiFC by including a 35S-driven RFP on the same backbone as the BiFC constructs, confirming our qualitative observations above (Fig. 2j). These results were not due to any mislocalization of the truncated versions of the CC1 as all the constructs still clearly labelled the plasma membrane (Fig. 2i-j, and Fig. S4f). From these analyses, we concluded that the N-terminal part of CC1 is necessary for the interaction of the protein with BEN1. The N-terminal part of CC1 consists of an intrinsically disordered region (IDR)29. Intriguingly, HopM1 (formerly hopPtoM) also interacts with BEN1 via an IDR in its N terminus, which promotes poly-ubiquitination and degradation of BEN127. Our results therefore highlight a related interaction mode and degradation scheme of BEN1 in relation of CC1 or HopM1. We speculate that CC1 and HopM1 either specifically recognize the poly-ubiquitinated BEN1, or recruits Ubiquitin ligase to BEN1 upon interaction, which in turn regulates the degradation of BEN1.
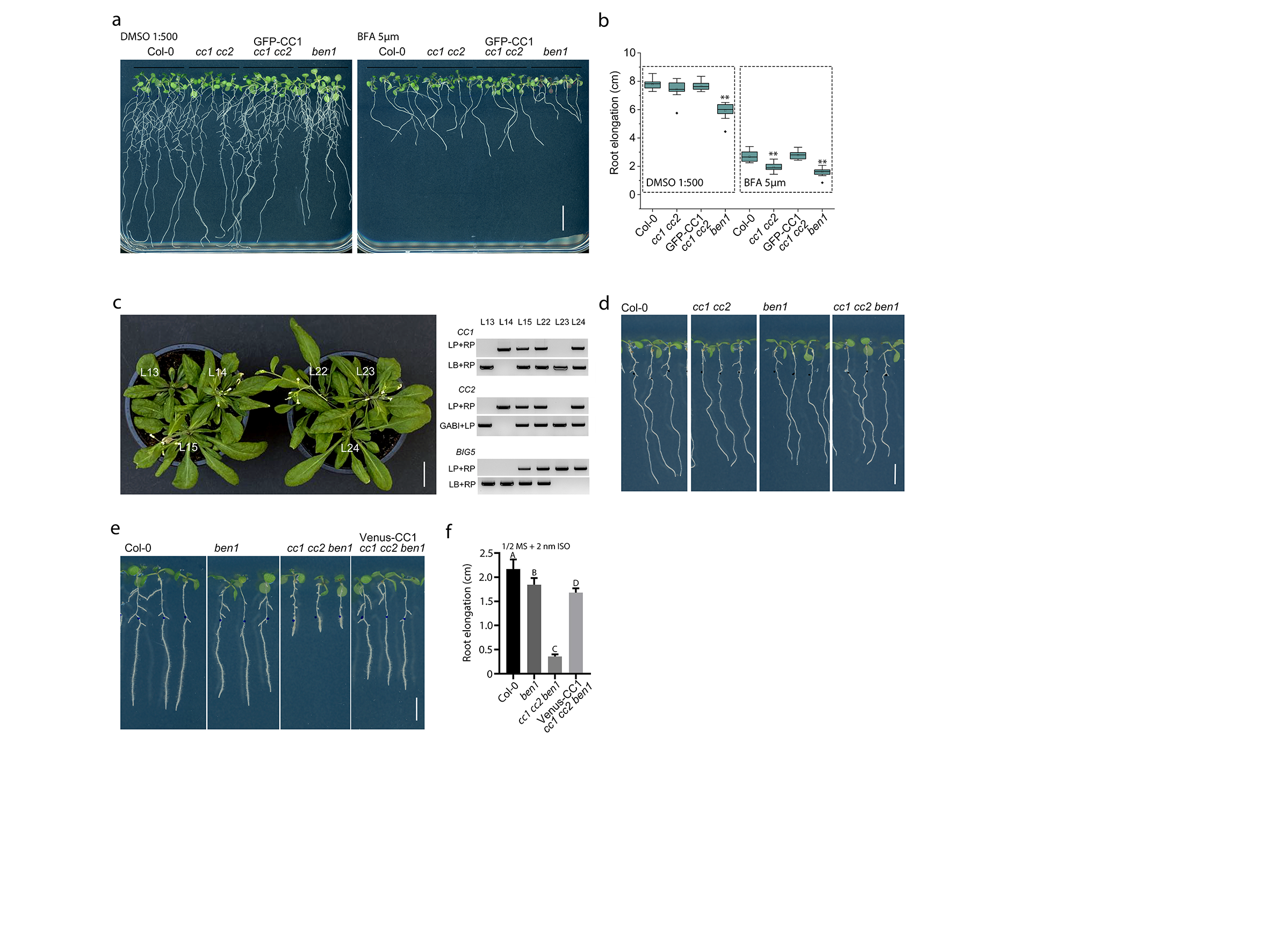
Given the general trafficking role of BEN1 at the TGN/EE, we next assessed how CC1 and BEN1 may functionally interact. We therefore grew ben1 and cc1cc2 mutant seedlings on media supplemented with the trafficking inhibitor BrefeldinA (BFA), a potent ARF-GEF inhibitor that leads to BFA-related endomembrane aggregates in the cytoplasm, also called BFA bodies30. Notably, cc1cc2 mutant seedlings were more sensitive to the BFA conditions than wild type and showed similar defects in root elongation as that of the ben1 mutant (Fig. S5a-b)31. We next generated ben1cc1cc2 triple mutants to investigate possible genetic interactions. Interestingly, ben1cc1cc2 triple mutant plants displayed reduced rosette leaf expansion, which corresponded to a lower level of crystalline cellulose, as compared to wild-type (Fig. 3a-b; S5c). In addition, ben1cc1cc2 seedlings displayed increased sensitivity to the cellulose synthesis inhibitor isoxaben (Fig. 3c-d; S5d).
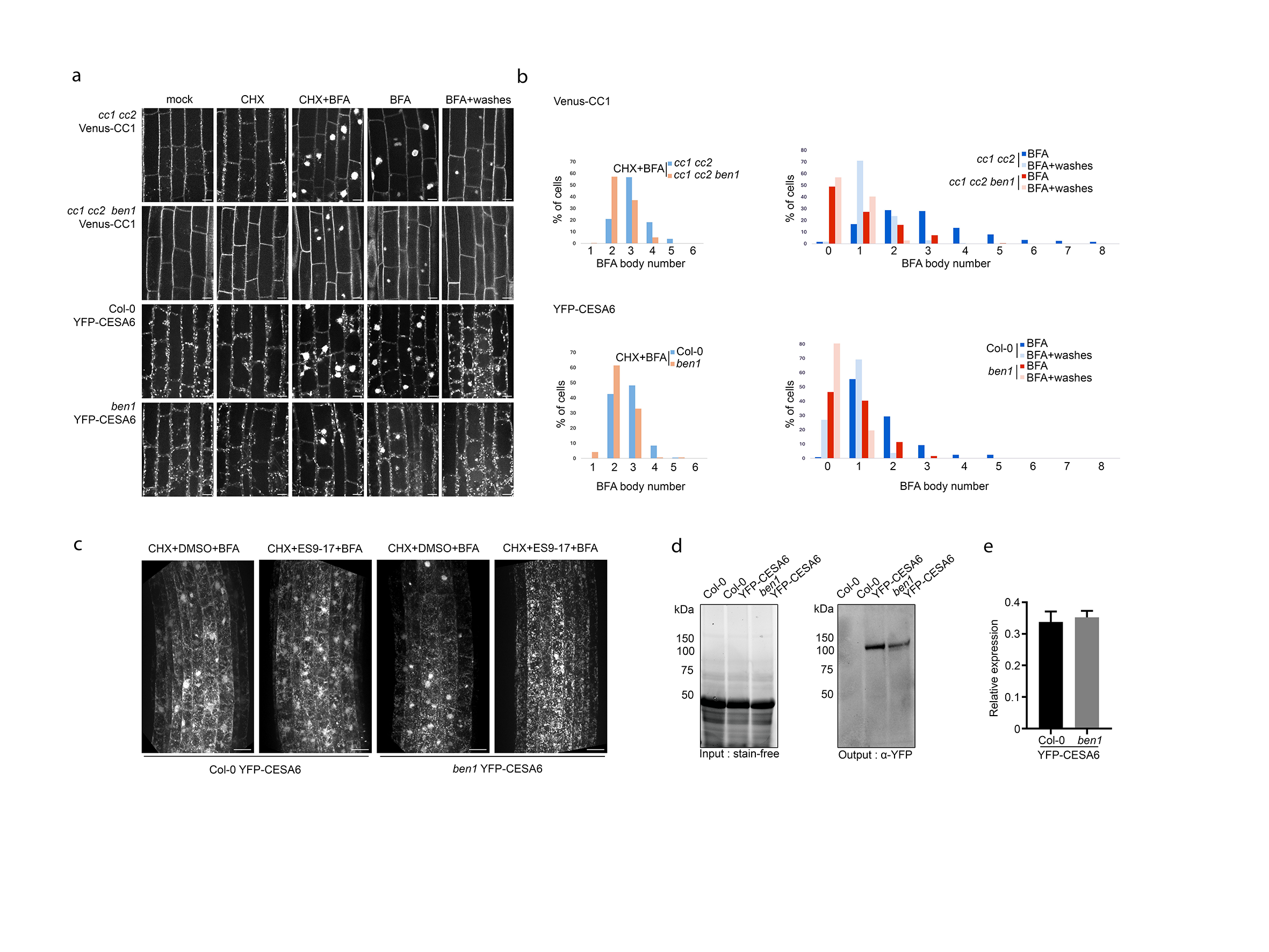
We next sought to investigate the cell biological relationship between CC1 and BEN1. To do this, we attempted to directly visualize the dynamic behavior of CC1 and the CSC in the ben1 mutant. We therefore introgressed Venus-CC1 and YFP-CESA6 into ben1 plants. Previous studies showed that mutations in BEN1 affect trafficking of, for example, PIN-FORMED (PIN) and BRASSINOSTEROID INSENSITIVE1 (BRI1), which led to reduced accumulation of the proteins in BFA bodies25, 32. Consistent with these reports, we observed reduced accumulation of both Venus-CC1 and YFP-CESA6 in BFA bodies in the ben1 mutant compared to the wild type (Fig. 3e-f). To investigate whether the BFA bodies were fueled from Endoplasmic Reticulum/Golgi (i.e. newly synthesized proteins en route to the TGN and plasma membrane), we pretreated seedlings with the de novo protein synthesis inhibitor cycloheximide (CHX), and then with BFA. These seedlings still contained substantial BFA bodies per cell (Fig. 3g; Fig. S6a-c), indicating that BEN1 mainly is associated with trafficking of the CSC between the plasma membrane and the TGN/EE. To assess if the CSCs were unable to get to the plasma membrane, or if there were problems in CSC internalization, in the ben1 mutant, we performed wash-out experiments of BFA. Here, the BFA bodies disappeared at similar rates in the ben1 mutant and WT, indicating that BEN1 may be related to internalization of the CSCs and not CSC trafficking from the TGN to the plasma membrane (Fig. S6a-b). Interestingly, the endocytosis of CSC is complex and may include both clathrin-dependent and independent processes15. To attempt to separate these processes in context of BEN1, we co-treated seedlings with the clathrin-mediated endocytosis inhibitor ES9-17 and CHX, and then with BFA. We found that this combination still led to BFA body formation, albeit to a substantially lesser degree than without ES9-17 (Fig. 3g-h, Fig. S6c). These results highlight the clathrin-independent internalization of the CSCs, and indicate that BEN1 may contribute both to clathrin-dependent and -independent endocytosis of the CSC (Fig.3g-h, Fig. S6c). Finally, we checked YFP-CESA6 protein abundance and mRNA levels in ben1 to see if the trafficking defects may also impact CESA6 levels. However, we did not find any major differences between ben1 and WT control (Fig. S6d-e). Taken together, our results indicate that BEN1 regulates the trafficking of the CSC between the plasma membrane and the TGN/EE and that defects in the BEN1 therefore impact cellulose synthesis.
Protein interactions are essential to understand how proteins work in context to each other and to infer protein function. The implementation of new tools to study PPIs is therefore of substantial interest across all aspects of cell biology. We provide a new toolbox for PPI inferences, PUP-IT, in plant biology and highlight how this tool may be used to identify new components of important processes in plants.
We show that PUP-IT may be used in a range of different plant biological systems, including cell suspensions, transient infiltration assays and in stable transgenic plants. In addition, we supply several vector constructs where both the bait and the substrate can be included on a single backbone. The substrate may be modified with different tags to enrich the labelled proteins, which increases the versatility of the toolbox. It is, however, important to realize that the tag needs to be placed upstream of the PUP, as the PafA recognizes a phosphorylated PUP to then ligate onto Lysine residues on proteins close to the PafA33, 34. We also compared PUP-IT against the highly popular TurboID. Here, we show that known interactors of TPLATE (used in the comparison as bait) were enriched to higher relative levels with PUP-IT as compared to TurboID and even against AP-MS. These results show that PUP-IT may have a higher specificity for close interactors, i.e. proteins forming protein complexes, than that of for example TurboID. We therefore envision that PUP-IT may become a preferred approach in the emerging field of protein proximity labelling.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}