Ethics and biosecurity protocols.
Ethics protocols approved for the experiments in mice ref. CEEAH 3603 and biosecurity protocols 345-16 and 407-17. All procedures were approved by the Committee of Ethics of the Universitat Autònoma de Barcelona and the Generalitat de Catalunya. They were also carried out in accordance with the European Communities Council Directive (2010-63-UE) and Spanish legislation (RD 53/2013).
Mice.
All experiments used male and naturally cycling female adult mice (2-6 months of age) housed in groups of 4 (except when mentioned) in a room with a 12:12h light/dark cycle (lights on from 8am to 8pm). All animals were housed with controlled temperature 22 ± 1 ºC and humidity (~ 40%). Behavioral procedures and pharmacological manipulations begun early in the light phase of the cycle. Tac2-Cre (stock# 018938 Jackson Labs) were bred within our animal facility. Wild-type C57BL/6J were purchased from Charles River (Barcelona, Spain). Male and female mice were housed separately in the same room.
Cued-Fear Conditioning.
For Fear Acquisition (FA) and Fear Expression (FE) test, a computerized Startle system was employed (Panlab-Harvard, Barcelona, Spain) as previously described (49). Delivery of tones and shocks was simultaneously controlled by Freezing v1.3.04 software (Panlab-Harvard, Barcelona, Spain). The fear chamber consisted of a black methacrylate box with a transparent front door (25x25x25cm) inside a sound-attenuating chamber (67x53x55cm). The same boxes were used for FA and FE.
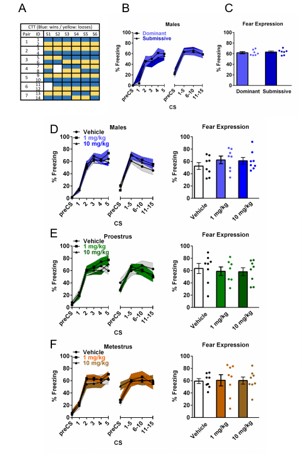
Animals were habituated to the chambers for 5 min/day during two consecutive days prior to FA. For cue-dependent fear conditioning, all animals remained 5 min in the fear chamber before the onset of the first tone. During FA, all groups received 5 trials consisting in a tone as the Conditioned Stimulus (CS) (30 s, 6 kHz, 75 dB) that coterminated with a footshock which served as the Unconditioned Stimulus (US) (1 s, 0.6 mA). The intertrial interval (ITI) was 3 min, and 3 additional min followed the last trial. All animals received the treatments 30 min after FA in order to manipulate the consolidation of memory and therefore avoid any effect during acquisition. The FE test was performed 24 hours after FA. Mice remained 5 min in the chamber before trials, and afterwards were exposed to 15 trials of the 30 s CS tone alone (cued-fear) with a 0.5 min of ITI interval. An additional 0.5 min interval followed the last trial of FE. Freezing behavior was used as an index of fear. The StartFear system allows recording and analysis of the signal generated by the animal movement through a high sensitivity weight transducer system.
To assure that freezing behavior was exclusive of the previous tone-shock conditioning, different contexts were utilized for FA and FE. FA context consisted of a yellow light source (~10 lux), a grid floor of 25 bars (3 mm Ø and 10 mm between bars) that dispensed the footshocks, a background noise of 60 dB produced by a ventilation fan and a solution of ethanol (EtOH) 70% (v/v) odor was used for cleaning between sessions. FE context consisted of a red light source (~10 lux), a grey floor covering the bars, no background noise and CR36 – bronopol 0.26 % (v/v), benzalkonium chloride 0.08% (v/v) and isopropyl alcohol 41% (v/v) – (José Collado, Barcelona, Spain) for cleaning, with changes in the length and turns of the transportation route from the vivarium to the testing room between FA and FE.
Confrontation Tube Test (CTT).
Male mice were pair-housed for 5 weeks before testing in order to establish stable social hierarchies between them. The confrontation tube consisted of a methacrylate tube (30 cm length, 3.6 cm inner Ø) with two lids at 13 cm from each end. All animals were habituated to the tube for 2 consecutive days before the test. The habituation consisted of 3 crossings from one end to the other of the tube and the tube was always cleaned with EtOH 70% (v/v) between animals. Mice were tested for 6 consecutive days, with 8 trials per day with ~20 min between trials. For each trial, each mouse from each couple entered the tube simultaneously by the extremes. When both animals reached the lids, these were lifted for animals to face each other and that’s when the test started. The first animal that put its four limbs outside the tube was considered the looser while the other one won the trial. When both animals remained 2 min inside the tube without confronting each other, the trial was considered null. The animal that presented more wins throughout each session was considered the winner of the session. At the end of the 6 sessions, the animal that presented more won sessions was considered dominant over its cage mate. The day after the 6th session of the tube test, animals underwent the FC protocol as abovementioned. All mice received osanetant 30 min after FA and were tested for memory recall 24 hours later to assess for differences in the effect of the drug between dominant and submissive mice.
50 uL of blood was collected from each animal by tail-nick two days before the first habituation to the tube, 2 hours before the first habituation session, 2 hours before the first test and 2 hours before the first habituation to the fear conditioning chamber, thus, with every two days since the first extraction. Blood was centrifuged (8000 g, 15 min, 4ºC) and serum was stored at -20ºC. Blood was not analyzed since we did not find behavioral differences between groups.
Vaginal smear cytologies.
In order to assess the phase of the estrous cycle that female mice presented during FA, all female mice were monitored for 3 to 4 consecutive cycles (approximately 10-14 days) before learning to test for regularity of the cycle. For that end, a 20 mL pipette was loaded with 10 mL of standard NaCl 0.9% (w/v) solution and later the tip was softly placed on the vaginal aperture. In case of urination when grabbing the animal, urine was cleaned using a regular tissue. The 10 mL of saline were unloaded and collected for 5 consecutive times in order to collect an enough amount cells for the assessment, and later placed on an adhesion slide (Superfrost Plus, Thermo Fisher, Barcelona, Spain). Slides were dried using a hot plate (HI1220, Leica, Madrid, Spain) at 37ºC for 30 min and later stained in Cresyl Violet Acetate (C5042, Sigma-Aldrich, Spain) 0.1% (v/v), washed twice for 1 min in distilled water and read in brightfield microscopy with a 10x or 20x objective in an Eclipse 80i microscope (Zeiss, Spain). Three different cell types may appear in the preparation: cornified epithelial cells, round nucleated epithelial cells or leukocytes. The different stages of the estrous cycle were assessed depending on the proportion of the abovementioned cells. Proestrus is characterized by a high proportion (>80%) of nucleated epithelial cells, that might present very small amounts of cornified epithelial cells or leukocytes. Estrus is typically presented with cornified epithelial cells with a lower grade of staining than leukocytes and nucleated epithelial cells. Metestrus presents a mixture of cornified epithelial cells and a considerable proportion of leukocytes. Diestrus is characterized by >90% of leukocytes that might present a very small proportion of round nucleated epithelial cells (see Fig S1E). After assessment of regular cycling, females were distributed in groups according to the stage of the estrous cycle they presented before FA, which is also the day they receive the different treatments after FA.
Drugs.
Intraperitoneal osanetant (Sigma-Aldrich, Spain) dose was 5 mg/kg (7), 1 mg/kg or 10 mg/kg, and intra-cerebral dose was 30 nmol per side as in our previous paper (7) using 0.1% Tween 20 in saline as vehicle. The dose of intra-Central Amygdala (CeA) Androgen Receptor (AR) agonist CI-4AS-1 (Tocris, UK) was 100 nM based on previous research showing decreased depolarization-induced suppression of excitation in POMC neurons (50), dissolved in 1% DMSO (v/v). We used the AR agonist instead of testosterone in order to avoid its possible conversion to 17-b-Estradiol by aromatase. The Akt activator SC 79 (Tocris, UK) was used at a dose of 0.1 µM in 1% DMSO (v/v) because it has shown to have a protective effect on dopaminergic neurons against oxidative stress (51). Anti-Akt1/2 (Tocris, UK) was used at 5 µM in 1% DMSO (v/v) due to its ability to block Akt phosphorylation (52). Estrogen Receptors (ERs) antagonist ICI 182,780 (Tocris, UK) was used at a 10 µM in 1% DMSO (v/v) as it has shown previously to be effective decreasing EtOH excitation of DA neurons in the VTA (53). CNO in 0.5% DMSO (v/v) systemically was dosed at 1mg/kg (7).
Surgery.
All surgeries were performed using isoflurane 5% (v/v) for induction, and 2-3% (v/v) for maintenance, in oxygen, at a constant rate of 1.5 L/min. After skin shave and skin disinfection with EtOH 95% (v/v) and iodine povidone 10% (v/v), ovariectomies were performed making a bilateral incision on the back of the animal, 1 cm lateral to the midline and right over the backlimbs line. Adipose tissue was extracted, and the ovary was localized and isolated making a knot with sterile absorbable suture thread (1019723, Centauro, Barcelona, Spain) around the oviduct. The ovary was extirpated and the adipose tissue, containing the rest of the oviduct, was returned to the abdominal cavity. Muscle was sewed with sterile absorbable thread and skin was sewed using sterile silk suture (1019717, Centauro, Barcelona, Spain). Mice remained resting for 6 weeks until trunk blood was collected to avoid any effect of previous estradiol.
For stereotaxic surgeries, after induction of sleep, mice were placed in the stereotaxic frame (Kopf Model 962, Harvard-Panlab, Barcelona, Spain). After alignment of the Antero-Posterior (AP) and Latero-Medial (LM) axis with the frame, injections of AAV8-hSyn-DIO-hM4D(Gi)-mCherry to the CeA the following coordinates were employed: AP -1.3, ML ± 2.5, Dorso-Ventral (DV) –4.4 mm from Bregma, as previously described (7). For cannulation, the same AP and ML coordinates were utilized, although cannulae were implanted at DV -3.4 mm due to the need to remain the CeA intact and because of the extra mm projection of the internal cannulae. Cannulae were secured to the skull with anchor screws for mice (Plastics One, Germany) and dental cement (Fortex Fájula, Cibertec, Spain) For further details on the virus and microinfusions, please see Adeno associate virus (AAV) infection and microinfusions section below.
Adeno-associate virus (AAV) infection and microinfusions.
We used AAV obtained from the pAAV-hSyn-DIO-hM4D(Gi)-mCherry (hM4di-mCherry DREADDs). The plasmid was obtained from Addgene and the AAV from the viral vector production unit at Universitat Autònoma de Barcelona. Serotype 8 of the AAV was bilaterally injected (0.5 mL/side) using a microinjection pump (1 mL per 15 min rate) into the CeA (coordinates abovementioned). Cannulae were placed using coordinates mentioned in the surgery section. For microinfusions of drugs in awake animals, internal cannulae projected 1 mm from the tip of the internal guide, reaching DV coordinate -4.4 mm from Bregma for injections, corresponding to the CeA. Bilateral injections were performed simultaneously and manually using two knurled hub, type 3 tip, 1 mL Hamilton syringe (external Ø 0.52 mm and internal Ø 0.26 mm) (Cibertec-Harvard, Madrid, Spain) coupled on one end to a polyethylene (PE) 50 tube (~ 20 cm) (Plastics One, Germany) with the internal cannula coupled to the other end of the tube. The PE-50 tube was filled with distilled water, leaving a 7.5 mm gap of air between the internal cannula and the water to avoid dilution of the drugs with water. Microinfusions were performed at a rate of 0.5 mL over 2 min and internal cannulae remained one extra min in place to prevent the backflow of the drug.
Steroids determination.
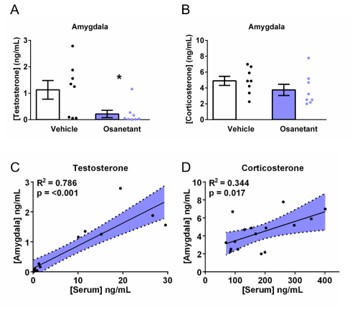
Trunk blood was collected after sacrificing the animals. Blood was centrifuged (8000 g, 15 min, 4 ºC). Serum was collected using a 200 µL pipette, transferred into a 2.5 mL Eppendorf tube and stored at -80 ºC for later analysis. Brains from animals euthanized 330 min after treatment (6 hours after FA) were snap frozen using Isopentane cooled with dry ice, and later stored at -80ºC until sectioning. Fresh frozen brains were then coronally sectioned until reaching a coronal plane 0.58 mm behind Bregma. 1 mm Ø micropunches from both Amygdalae were extracted reaching coronal plane 1.94 mm behind Bregma, transferred into a 2.5 mL Eppendorf tube and stored until processing. 400 mL of Formic Acid (F0507, Sigma-Aldrich, Spain) 0.1% (v/v) were added to each tube and sonicated for consecutive five cycles. The sonication probe was then rinsed with 800 mL of Acetonitrile (271004, Sigma-Aldrich, Spain) that were added to the tubes, and these were vortexed for 2 s. Micropunches extracts were stored at -80ºC until analysis. Both plasmatic and amygdala levels of testosterone, progesterone, dehydrocorticosterone and deoxycorticosterone were determined based on previously reported papers (54, 55). Seric and amygdalar testosterone, progesterone, dehydrocorticosterone and deoxycorticosterone were evaluated were evaluated based on previously reported papers (54, 55). Briefly, 20 µL of plasma were mixed with 20 µL of labelled internal standard solution. After proteins precipitation with 100 µL of acetonitrile, samples were centrifuged (3000 g, 5 min) and the supernatant transferred to a clean tube. In the case of micropunches 20 µL of labelled internal standard solution was added to the 1 mL of extract from micropunches. The mixture was vortexed and transferred to a clean tube. Both plasma and micropunches underwent a liquid-liquid extraction by addition of 1 mL of NaCl (saturated solution) and 4 mL of ethyl acetate. Extracts were centrifuged (3000 g, 5 min) and the organic layer was transferred into a clean tube and dried under a nitrogen stream. Dried extracts were reconstituted with 100 µL of methanol and 10 µL were injected into the LC-MS/MS system consisting on an Acquity UPLC system coupled to a triple quadrupole (TQS Micro) mass spectrometer. Steroids detection was performed by selected reaction monitoring (SRM) including 2 transitions for each analyte. The most specific one was selected for the quantification. Quantification was performed by external calibration approach using the TargetLynx module of the MassLynx software (Thermo Fisher, Barcelona, Spain). Estradiol was measured with the ELISA kit ES180S-100 (Calbiotech, California, USA). For standard curve preparation, serum from ovariectomized mice was used adding known concentrations of estradiol (E885-250MG, Sigma-Aldrich, Spain) -0, 3, 10, 30, 100 and 300 rg/mL- utilizing denaturalized EtOH (Casa Álvarez, Barcelona, Spain) as a vector for estradiol. Kit instructions were followed as stated, samples were loaded in duplicates and absorbance was read at 450 nm with the microplate reader Varioskan Lux (Thermo Fisher, Barcelona Spain) controlled with Skanit for microplates v6.0 software (Thermo Fisher, Barcelona, Spain). Average of duplicates was used as estradiol determinations for each sample.
Immunohistochemistry.
For immunohistochemistry assays, mice were transcardially perfused with 50 mL of 4% (v/v) paraformaldehyde (PFA) (Casa Álvarez, Barcelona, Spain) for 5 to 6 min, then decapitated and brains were extracted and stored in 4% (v/v) PFA for 24 hours. After this time, brains were rinsed (3 times, 10 min each) with 1x Sorenson’s PB consisting of 10.9 g/L Sodium phosphate dibasic (0876, Sigma-Aldrich, Spain), 3.2 g/L Sodium phosphate monobasic (04270, Sigma-Aldrich, Spain) and transferred into 30% Sucrose (84097, Sigma-Aldrich, Spain) in 1x Sorenson’s PB in conic Falcon tubes (Thermo Fisher, Barcelona, Spain) until the brain reached the bottom of the tube (~ 48 to 72 hours). Right after, brains were snap frozen in a metal cube containing Isopentane (M32631, Sigma-Aldrich, Spain) cooled with dry ice and stored at -80ºC until sectioning.
For Neurokinin 3 Receptor (Nk3R), Glutamic Acid Decarboxylase 65 (GAD65), Calmoduline Kinase II a (CaMKIIa) and vesicular Glutamate Transporter 2 (vGLUT2) fluorescent immunostaining brain sections (30 mm/section) were rinsed 3 consecutive times with 1x KPBS. All incubations were performed on top of a shaking platform. Right after washing the slices, these were incubated for 60 min in blocking buffer (5% Donkey Serum and 0.4% Triton-X in 1x KPBS) at 4 ºC. After incubation in blocking buffer, sections were incubated overnight with the following primary antibodies: rabbit anti-Nk3R (Donated by Philip Cioffi, INSERM, 1:2500), chicken anti-GAD65 (139958, Abcam, 1:500), goat anti-CaMKIIa (87597, Abcam, 1:500) and mouse monoclonal anti-vGlut2 (ab79157, Abcam, 1:300). Primary antibodies were diluted in 0.4% Triton-X in 1x KPBS. After incubation in primary antibodies solution, brain slices were rinsed 3 times with KPBS 1x and then incubated in a sary antibodies solution for 2 hours at room temperature. The sary antibodies solution was prepared in Triton X (x100, Sigma-Aldrich, Spain) 0.4% (v/v) in 1x KPBS, and included the following sary antibodies conjugated to fluorophores: donkey anti-rabbit AlexaFluor488 (115-546-072, Jackson Immunoresearch, 1:1000), donkey anti-chicken Cyanine3 (703-166-155, Jackson Immunoresearch, 1:1000), donkey anti-goat AlexaFluor594 (705-586-147, Jackson Immunoresearch, 1:1000) and donkey anti-mouse AlexaFluor647 (715-606-150, Jackson Immunoresearch, 1:1000). 4′,6-diamidino-2-phenylindole (DAPI) (10236276001, Sigma-Aldrich, 1:10000) was used to stain cell nuclei.
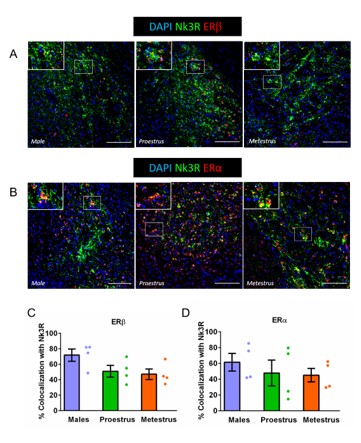
Z-Stacks of the Central Amygdala (0.50 μm/interval) were acquired using a Leica SP5 confocal microscope (Leica, Spain) with a PL APO 40x/1.25-0.75 immersion objective. Colocalization analyses were performed using Fiji for Windows. Z-Projections of Nk3R signal were used to create a mask. This mask was then applied to Z-Projections (0.50 μm/interval) of GAD65, CaMKIIa and vGLUT2 stacks using LungJ plugin. Mander’s colocalization coefficient of colocalization between Nk3R and GAD65, CaMKIIa and vGLUT2 was calculated using Just Another Colocalization Plugin (JACoP). Average colocalization index between right and left CeA was used a measure of colocalization for each animal.
For Estrogen Receptor b (ERb) and Estrogen Receptor a (ERa) fluorescent immunostaining, the staining protocol was similar to the one mentioned above. Primary antibody solution contained rabbit anti-Nk3R (Donated by Philip Cioffi, 1:2500) and mouse monoclonal anti- ERa (sc-8002, Santa Cruz, 1:50) or mouse monoclonal anti- ERb (288, Abcam, 1:500). Sary antibodies solution included donkey anti-rabbit AlexaFluor488 (115-546-072, Jackson Immunoresearch, 1:1000) and donkey anti-mouse Rhodamine Red-X (715-295-150, Jackson Immunoresearch, 1:1000). DAPI (Sigma-Aldrich #10236276001) was used to stain cell nuclei. Z-Stacks of the Central Amygdala (0.50 μm/interval) are acquired using a Zeiss LSM 700 confocal microscope (Zeiss, Spain) with a PL APO 40x/1.25-0.75 immersion objective. Cell counter plugin for Fiji use employed to measure the number of Nk3R+ cells in the CeM. Mander’s colocalization coefficients for ERa and ERb were calculated as abovementioned for Nk3R, GAD65, CaMKIIa and vGLUT2 immunohistochemistry using JACoP plugin in Fiji for Windows.
Cannula placement verification.
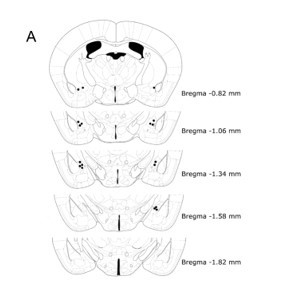
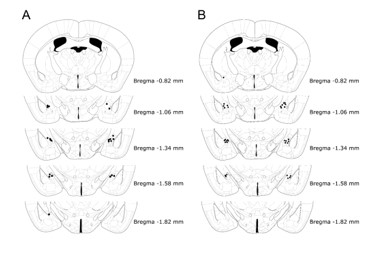
For cannula placement verification, amygdala brain sections were stained with a standard Nissl staining protocol. Brains were sectioned (30 μm/section) using a Leica Cryostat (-20 ºC for the chamber, -18 ºC for the sample), direct to mount and stored at -20 ºC until staining. Slides were dried on a hot plate at 37 ºC overnight before staining. First, slides were dehydrated in consecutive decreasing concentrations of denaturalized EtOH (Casa Álvarez, Barcelona, Spain): 3 min in EtOH 100% (v/v), 3 min in EtOH 95% (v/v) and 3 min in 70% (v/v). Right after that, slides were rinsed in distilled water three times for 2 min each to extract any trace of sucrose. Samples were then stained for 8 min in a Cresyl Violet Acetate Solution (C5042) consisting of 1 mg/mL Cresyl Violet Acetate in Walpole Solution, prepared with Glacial Acetic Acid (S2889, Sigma-Aldrich, Spain) 60% (v/v) and Sodium Acetate 0.2M (S2889, Sigma-Aldrich, Spain) 40% (v/v). After staining, slides are rinsed twice in distilled water for 2 min each and then dehydrated using increasing concentrations of EtOH: 3 dips in EtOH 50% (v/v), 3 dips in EtOH 70% (v/v), 3 dips in EtOH 96% (v/v) and 3 min in EtOH 100% (v/v). After this last dehydration, slides are incubated in Xylene (214736, Sigma-Aldrich, Spain) 3 times for 3 min each and then covered using DPX mountant for histology (06522, Sigma-Aldrich, Spain). Slides remain untouched for 24 hours the dark until stored in a regular box for slides at room temperature.
Cannulae placement verification was performed using brightfield photographs obtained using a 10x objective in an Eclipse 80i microscope (Leica, Spain). Animals that did not present a bilateral cannula lesion in the central amygdala were discarded from experiments.
mRNA qPCR array.
30 min after receiving osanetant (1 hour after FA), animals were decapitated, and brains were immediately fresh frozen in isopentane cooled with dry ice and stored at -80 °C. Amygdala tissue from both hemispheres was extracted by 1mm micropunch as previously described and each structure from each mouse was individually stored. Total RNA was isolated and purified from the tissue with the RNeasy Mini Kit (74106, Qiagen) following the manufacturer’s instructions. Total RNA was isolated with Maxwell RSC simplyRNA Tissue Kit (AS1390, Promega). Quantus Fluorometer (E6150 Promega) was used to ensure the quality of RNA before the qPCR array (PAMM-071ZC-24, Qiagen). Gusb was the housekeeping gene use for normalization of qPCR results. qPCR array was performed following the instructions of the kit as stated, using the thermocycler 7500FAST, controlled by 7500FAST v2.0.6 software both from Applied Biosystems (Thermo Fisher, Barcelona, Spain).
Ingenuity Pathway Analysis – Bioinformatics.
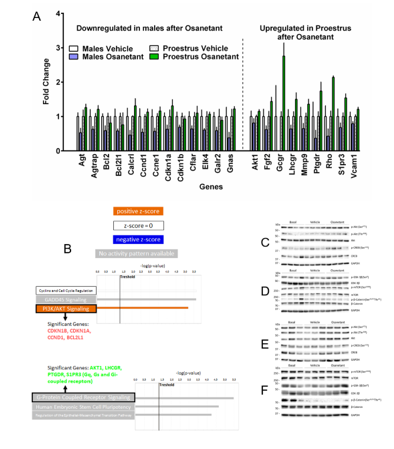
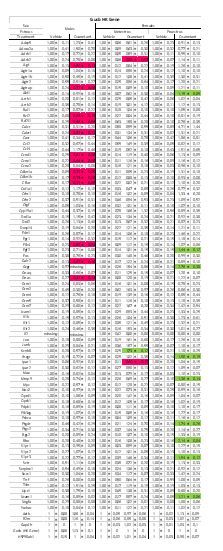
The gene list obtained on the mRNA qPCR array was analyzed with Bioinformatics. We used Ingenuity IPA version #44691306. The male and female lists of genes selected according to their significant p-value were uploaded separately. All genes were introduced with their respective expression fold change value. The female’s gene list contained the genes Akt1, Gcgr, Fgf2, Lhcgr, Mmp9, Ptgdr, Rho, S1pr3, Vcam1. The males’ gene list contained the genes Agt, Agtrap, Bcl2, Calcrl, Ccnd1, Ccne1, Cdkn1a, Cdkn1b, Cflar, Elk4, Galr2, Gnas, Bcl2l1. We performed a Core Analysis/Expression Analysis using the fold change as a measurement to base the analysis on. These analyses were performed separately for males and females by using most of the predetermined criteria by IPA. The general settings of this analysis included both direct and indirect relationships based on the references set “Ingenuity Knowledge Base (Genes Only)”. For the networks analysis we selected 35 molecules per network and 25 networks per analysis. We also choose all Node Type and all Data Sources. For Confidence we selected only “Experimentally observed”. For Species we selected all. The results we obtained with this IPA analysis were focused in two categories: Canonical Pathways (Fig 3) and Upstream analysis (Fig S5). For the Canonical Pathways we customized the charts by using a criterion of only selecting the categories that presented more than 4.5-fold changes in both males and females. The graphic in Fig 3 was created opening the PI3K/Akt in males and the G-coupled protein receptor signaling in females. Both graphics were combined using the Path Designer tool.
Biochemical studies.
10 min after receiving osanetant (40 min after FA), male and proestral female mice were decapitated and brains were snap frozen in isopentane right before storage at -80ºC. Both amygdalae were microdissected as abovementioned with a 1 mm micropunch, homogenized by sonication in 80 µL of cold-lysis buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% Na-deoxycholate, 2.5 mM EDTA, 1 mM Na3VO4, 25 mM NaF) containing protease and phosphatase inhibitors (Roche España, Barcelona, Spain). Protein concentration was quantified with the BCA protein assay kit (Thermo Fisher Scientific, Barcelona, Spain), resolved on SDS-polyacrylamide gel electrophoresis and detected by Western blotting with the following antibodies: rabbit anti-phosphorylated Akt (Thr308, 1:1000; Cell Signaling; Ser743, 1:1000; Cell Signaling) and goat anti-total Akt (1:1000; Santa Cruz Biotechnology), rabbit anti-CREB (1:700; Cell Signaling) and anti-phosphorylated CREB (Ser133; 1:1500; Cell Signaling), rabbit mTOR (1:1000; Cell Signaling) and anti-phosphorylated mTOR (Ser2448; 1:1000; Cell Signaling), mouse GSK3β (1:2500; BD Biosciences) and rabbit anti-phosphorylated GSK3β (Ser9; 1:1000; Cell Signaling), rabbit anti-β-Catenin (1:6000; Sigma-Aldrich) and anti-phosphorylated β-Catenin (Ser33/37/Thr41; 1:1000; Cell Signaling) or mouse anti-GAPDH (1:100000; Life Technology). Then, protein bands were detected with sary antibodies coupled to peroxidase enzyme (Bio-Rad, Madrid, Spain) and enhanced chemiluminescent reagent were captured in ChemiDoc MP System (Bio-Rad, Madrid, Spain) and quantified in a linear range using the ImageLab v5.2.1 software (Bio-Rad, Madrid, Spain) as reported (56).
Data analyses.
Statistics analyses were performed using IBM SPSS Statistics 23.0. Detection of outliers was using the Grubb’s test and removed when it was appropriate. For experiments involving independent samples, when these presented normal distributions and equality of variances, one-way ANOVA (GLM) was employed; otherwise non-parametric analyses were utilized for one factor analyses. Wald’s c2 with pairwise comparisons was used in Generalized Linear Model for multifactorial analyses that did not accomplish normality or homocedasticity. Additional individual comparisons were performed when appropriated.
For related sample analyses, repeated measures ANOVA was performed, using trials for FA or mean of groups of 5 trials for FE test. Equality of variances and sphericity were tested. When sphericity could not be assumed, Greenhouse-Geisser statistics were used for assessing significance and the corrected degrees of freedom were provided. ANOVA was used for evaluation of simple main effects and when interaction between main effects was significant. The results are presented as means ± or + SEM, and statistical significance was set at P ≤ 0.05.
Normality and homocedasticity tests, as well as statistics with main effects, interactions, pairwise analyses and effect size, when appropriate, are represented in Dataset S1. In and Dataset S2, 95% confidence intervals are represented.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}