Geometry optimization for 2-(2-hydroxy-5-methoxybenzylidene)hydrazinecarbothioamide (HMHC) and 2-(2-hydroxy-5methoxybenzylidene)-N-methylhydrazinecarbothioamide (HMNHC) was attempted using DFT/B3LYP/6-311 + + G(d,p) formalism. The computations identified the existence of both intra-molecular and bifurcated intra-molecular hydrogen bonds in both the molecules. Time-dependent density functional theory (TD-DFT) was employed to simulate Ultra-Violet spectra for both HMHC and HMNHC in order to substantiate experimental spectra in a solution of dimethyl formamide. For the two compounds under investigation, specific global reactivity descriptors were estimated with the help of frontier molecular orbital (FMO) analysis to understand the origin of UV-Vis spectra. Nonlinear optical (NLO) profile, for each of the two molecules, was computed, with in the frame work of the DFT/B3LYP/6-311 + + G(d,p) formalism to determine their utility for NLO applications. NBO analysis of hyper conjugate interactions made it possible to interpret the molecules NLO behavior in terms of intramolecular charge transfer (ICT). The reactive sites around the molecules were identified using Fukui function investigations and Molecular Electrostatic Potential (MEP).
Research Article
Quantum Chemical Investigation of (E)-2-(2-hydroxy-5- methoxybenzylidene)hydrazinecarbothioamide and its N-methyl Variant by Using DFT Methods
https://doi.org/10.21203/rs.3.rs-3881706/v1
This work is licensed under a CC BY 4.0 License
Version 1
posted
You are reading this latest preprint version
Thiosemicarbazones
DFT
UV-Visible spectra
FMO
NBO
NLO
- Bifurcated hydrogen bond and intra-molecular hydrogen bond exist in both HMHC and HMNHC molecules
- The two compounds undergo UV-Visible transitions, of the type n→π* and π→ π*
- The molecules are highly stable and reactive.
- HMHC and HMNHC have potential applications as NLO materials.
- The area around oxygen atoms of the hydroxyl moiety and methoxy groups is the best location for an electrophilic assault.
Heterocyclic thiosemicarbazones are a class of thiourea derivatives that have been the subject of extensive research because of their wide range of biological activities, widespread industrial use, and analytical sensitivity to different metal ions. Since 1956, when Brockman et al [1]. discovered the antitumoral property of 2-formylpyridine thiosemicarbazone derivatives, they have been used in biological applications. This inspired intense research investigations involving, numerous thiosemicarbazone derivatives for development of antibacterial, antiviral, antifungal, antioxidant, anti-inflammatory, and antiproliferative medicines [2–5]. Additionally, the effectiveness of this class of drugs against trypanosomiasis, coccidiosis, psoriasis, leprosy, psoriatic arthritis, and tuberculosis has been investigated [6, 7]. Herpes simplex virus (HSV) infection was selectively inhibited by several thiosemicarbazones both in vitro and in vivo. Impact of thiosemicarbazones on the human immunodeficiency viruses (HIV) has been documented [8]. It was found out that the biological activity of the thiosemicarbazones made from 2-formylpyridine and 2-acetylpyridine was increased by the presence of a bulky substitution at the N-terminal nitrogen atom [9–11]. Further, anticancer properties of thiosemicarbazones were attributed to their ability to inhibit ribonucleotide reductase, the enzyme that catalyzes the rate-limiting stage of DNA synthesis [12, 13]. The 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) [14] and Di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone [15], were subjects of extensive research to demonstrate their utility as medicine. The possible biological activity of the thiosemicarbazones is expected to be caused by the donor atoms of nitrogen and sulfur present in the compound [16]. Synthesis, and crystal structure, along with antioxidant, non-haemolysis and anti-inflammatory properties of 2-(2-hydroxy-5-methoxybenzylidene)hydrazinecarbothioamide (HMHC) and 2-(2-hydroxy-5-methoxybenzylidene)-N-methylhydrazinecarbothioamide (HMNHC) are available [17]. Hence we undertook this theoretical work with regard to their equilibrium geometry, UV-Visible spectra, frontier molecular orbital (FMO) characteristics, non-linear optical (NLO) behaviour, electrostatic potential surface (MESP) and natural bond orbital (NBO) properties. The purpose was: to demonstrate how far the theoretical parameters evaluated for HMHC and HMNHC agree with their corresponding available experimental counterparts [17–19].
All results presented in this article were accomplished by using Gaussian 09W/DFT program suit [21]. Important components that were used together for calculations consisted of (a) Becke’s three-parameter hybrid exchange functional B3;[22] (b) Lee-Young-Parr gradient corrected correlation functional[23] and (c) the split valence basis set, 6-311 + + G(d, p) expanded by including d-polarization functions on heavy atoms (carbon, oxygen, sulfur) and p-polarization functions on hydrogen atoms to achieve better description for polar bonds [24–26]. This is indicated as DFT/B3LYP/6-311 + + G(d,p) formalism. In order to start geometry optimization, one needs the initial structure of the molecule of interest. Fortunately, X-ray structure is available17 for HMHC and HMNHC. They were taken as the initial structure for geometry optimization. This was subjected to a rigorous geometry optimization by making simultaneous relaxation of all structural parameters. This procedure gave a non-planar structure of C1 symmetry for both HMHC and HMNHC. The authenticity of optimized structure was ascertained by the absence of imaginary frequencies. By using time-dependent density functional theory (TD-DFT), electronic absorption spectra of HMHC and HMNHC were simulated, in a solution of dimethyl formamide (DMF) which was used as a solvent to record UV-Vis spectra of both the compounds, by earlier workers [17]. Effect of the solvent was addressed by means of polarizable continuum model, employing an updated version of integrated equation formalism (IEF-PCM) [27]. This is a part of Gaussian 09 program suit.
Frontier molecular orbital (FMO) parameters, comprising of orbital energy gap (ΔE = EHOMO-ELUMO), ionization potential (I=-EHOMO), electron affinity (A=-ELUMO), global chemical softness (S = 1/(2η)), electronegativity (χ=(1 + A)/2), chemical potential(µ=-(I + A)/2), global electrophilicity index (ω = µ2/2η), and maximum charge transfer index (ΔNmax=-µ/η) [28–31], were calculated for both HMHC and HMNHC. Here HOMO and LUMO are the frontier molecular orbitals, known as highest occupied molecular orbital and the lowest unoccupied molecular orbital, respectively. Further, EHOMO and ELUMO are the energies of HOMO and LUMO orbitals, respectively. Non-linear optical behavior of a substance is defined by µt and its components, αt and its components, ∆α and βt, where µt, αt, ∆α and βt are its total dipole moment, total molecular polarizability, anisotropy of polarizability, and total first-order hyperpolarizability, respectively. All these parameters were computed for both molecules HMHC and HMNHC, using required equations, and employing DFT theory in combination with Buckingham’s formalism based on finite field approach [29].
The main structural aspect of HMHC and HMNHC is the π-electron delocalization between the mequinol ring and side chain in which lone pairs of two oxygen atoms, three nitrogen atoms, and one sulfur atom are also involved. This characteristic needs to be quantified, which requires understanding of second order interactions between occupied orbitals of one subsystem and empty orbitals of another subsystem. Therefore, an estimate of donor-acceptor interactions was made, based on second order perturbation analysis of Fock matrices in NBO basis for both HMHC and HMNHC, employing NBO 3.1 computer program suit [32], which is available with the Gaussian 09/DFT software suit. The exact equation for molecular electrostatic potential V(r) at a point positioned at r, due to the nucleus A with charge ZA situated near RA and electron charge density function ρ(rˈ) located at rˈ is given by [28, 33].
\(V(\mathbf{r})=\sum\limits_{A} {\frac{{{Z_A}}}{{|{\mathbf{R}_A} - \mathbf{r}|}}} - \int {\frac{{\rho (\mathbf{r}^{\prime})dr^{\prime}}}{{|\mathbf{r}^{\prime} - \mathbf{r}|}}}\)
where, ZA, r, RA and rˈ are taken with respect to an arbitrary origin.
In the above mentioned expression, the negative interaction energy is caused by the interaction of electrons with a positive test charge at ‘r’ as indicated by the second term on the right side of the expression, whereas the positive interaction energy arises due to interaction of the nuclei of the molecule with the same positive test charge used above at ‘r’ as indicated by the first term on the right hand side of the expression. Molecular electrostatic potential surfaces and associated electron density for HMHC and HMNHC were calculated by mapping MESP onto iso-electron density surfaces, at the level of DFT/B3LYP/6-311 + + G (d, p) formalism.
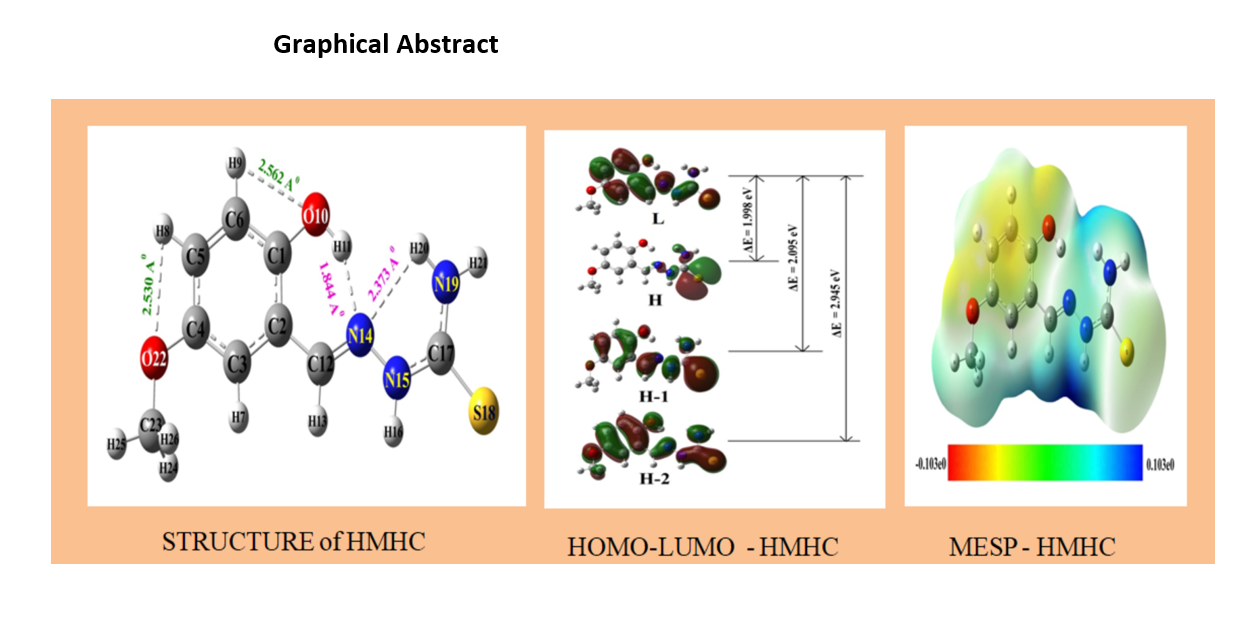
3.1 Optimized molecular geometry and bifurcated intra-molecular hydrogen bond
Equilibrium molecular geometry of the two molecules under investigation is decided by two forces, that oppose each other:
(i) Conjugation between the π-electron cloud of the mequinol ring and lone pairs on the three nitrogen atoms (N14, N15, and N19) along with lone pair electrons on the two oxygen atoms (O10 and O22) and lone pair electron on the sulfur atom tend to give coplanar structure to the molecule, which is further strengthened by the bifurcated intra-molecular hydrogen bond between N14 and (H11, H20) atoms, and intra-molecular hydrogen bonds between O22 and H8; and O10 and H9;
(ii) The steric repulsion effects in each of the molecules, that tend to stabilize the molecule in non-planar conformation, so as to minimize the steric effect. Primary source of this effect is the steric forces between pairs of hydrogen atoms (H7, H13) (H11, H20) and (H13, H16) along with that between (H16, S18). The most stable geometry of the molecules is dictated by the equilibrium between the above mentioned two opposing factors. The relative strength of the effects gets reflected in the dihedral angles associated with flexible bonds (C1-O10, C4-O22, O22-C23, C2-C12, N14-N15, N15-C17 and C17-N19). This delicate balance is significantly affected by two theoretical factors:
(i) Amount of correlation energy incorporated into computations, by theoretical method, and
(ii) The size of the basis set utilized for the computations.
Iterative method was implemented to solve self-consistent field equations, so as to obtain optimized molecular structure for both HMHC and HMNHC. The resulting structures are depicted in Fig.1. Optimized geometry structure data comprising of bond lengths, bond angles and torsional angles are collected in Table 1. Bifurcated intra-molecular hydrogen bond parameters are presented in Table 2.
Table 1. Experimental and DFT/B3LYP/6-311 ++ G(d,p) optimized geometry parameters of HMHC and HMNHC.
|
Atoms with numbering |
HMHC Molecule |
HMNHC Molecule |
|||||||
|
Bond Length (in Å) |
Bond Angle ( in °) |
Dihedral Angle ( in ° ) |
Bond Length (in Å) |
Bond Angle ( in °) |
Dihedral Angle ( in °) |
||||
|
C1 |
- |
- |
- |
- |
- |
- |
|||
|
C2 |
C1 |
1.410 (1.388) |
- |
- |
1.409 (1.391) |
- |
- |
||
|
C3 |
C2 |
C1 |
1.414 (1.401) |
119.19 (119.42) |
- |
1.414 (1.410) |
119.11 (118.80) |
- |
|
|
C4 |
C3 |
C2 |
C1 |
1.386 (1.377) |
120.82 (120.50) |
-0.35 (-0.641) |
1.386 (1.372) |
120.85 (120.76) |
-0.33 (-0.20) |
|
C5 |
C4 |
C3 |
C2 |
1.404 (1.382) |
119.22 (119.15) |
0.21 (1.161) |
1.404 (1.384) |
119.25 (119.41) |
0.19 (0.06) |
|
C6 |
C5 |
C4 |
C3 |
1.382 (1.372) |
120.66 (121.17) |
0.06 (0.790) |
1.382 (1.370) |
120.61 (120.64) |
0.05 (0.70) |
|
H7 |
C3 |
C2 |
C1 |
1.082 0.930 |
118.19 (119.78) |
179.77 (179.30) |
1.082 (0.930) |
118.20 (119.59) |
179.77 (179.73) |
|
H8 |
C5 |
C4 |
C3 |
1.083 (0.930) |
118.49 (119.43) |
179.95 (179.24) |
1.083 (0.930) |
118.52 (119.68) |
179.97 (179.30) |
|
H9 |
C6 |
C5 |
C4 |
1.083 (0.930) |
120.89 (119.95) |
179.78 (179.86) |
1.083 (0.930) |
120.89 (119.72) |
179.82 (178.47) |
|
O10 |
C1 |
C6 |
C5 |
1.354 (1.365) |
118.05 (117.92) |
179.84 (179.58) |
1.355 (1.368) |
118.06 (118.06) |
179.84 (179.40) |
|
H11 |
O10 |
C1 |
C6 |
0.975 (0.820) |
107.81 (109.49) |
-178.37 (-179.63) |
0.975 (0.819) |
107.72 (109.41) |
178.53 (171.70) |
|
C12 |
C2 |
C1 |
O10 |
1.449 (1.450) |
122.53 |
0.05 (0.272) |
1.450 (1.444) |
122.64 (122.24) |
-0.004 (-2.70) |
|
H13 |
C12 |
C2 |
C1 |
1.094 (0.930) |
116.71 (118.59) |
-176.31 (-178.89) |
1.094 (0.930) |
116.65 (118.13) |
176.54 (177.57) |
|
N14 |
C12 |
C2 |
C1 |
1.292 (1.279) |
123.34 |
3.46 (1.123) |
1.292 (1.284) |
123.511 (121.40) |
-3.20 (-2.44) |
|
N15 |
N14 |
C12 |
C2 |
1.361 (1.380) |
117.50 (115.03) |
-178.56 (-178.88) |
1.359 (1.381) |
117.58 (114.61) |
178.50 (173.32) |
|
H16 |
N15 |
N14 |
C12 |
1.015 (0.860) |
120.73 (119.43) |
-5.03 (-2.97) |
1.015 (0.859) |
120.73 (119.34) |
-4.33 (-4.88) |
|
C17 |
N15 |
N14 |
C12 |
1.370 (1.339) |
123.49 (121.10) |
-176.25 (-176.99) |
1.377 (1.358) |
123.75 (121.41) |
176.32 (175.08) |
|
S18 |
C17 |
N15 |
N14 |
1.674 (1.687) |
119.78 (118.81) |
-177.67 (-173.98) |
1.677 (1.676) |
118.97 (117.19) |
177.73 (166.30) |
|
N19 |
C17 |
N15 |
N14 |
1.348 (1.313) |
115.99 (118.89) |
1.89 (7.24) |
1.344 (1.319) |
115.84 (117.46) |
-2.37 (-13.28) |
|
H20 |
N19 |
C17 |
N15 |
1.007 (0.860) |
121.54 (120.01) |
8.21 (0.01) |
1.008 (0.860) |
117.61 (117.79) |
-3.19 (-0.46) |
|
H21 |
N19 |
C17 |
N15 |
1.005 |
117.58 (119.99) |
176.22 (19.99) |
- |
- |
- |
|
C21 |
N19 |
C17 |
N15 |
- |
- |
- |
1.455 (1.446) |
123.62 (124.32) |
179.83 (179.59) |
|
O22 |
C4 |
C3 |
C2 |
1.367 (1.373) |
125.06 |
-179.82 (-179.17) |
1.367 (1.366) |
125.04 124.35() |
179.82 (179.08) |
|
C23 |
O22 |
C4 |
C3 |
1.419 (1.419) |
118.35 |
0.33 (0.120) |
1.419 (1.414) |
118.36 (118.21) |
-0.14 (-0.46) |
|
H24 |
C23 |
O22 |
C4 |
1.096 (0.960) |
111.44 |
-61.60 (-61.78) |
1.096 (0.960) |
111.43
|
-61.11 (-59.58) |
|
H25 |
C23 |
O22 |
C4 |
1.088 (0.960) |
105.93 |
179.69 (178.25) |
1.088 (0.960) |
105.93
|
-179.83 (-179.6) |
|
H26 |
C23 |
O22 |
C4 |
1.095 (0.960) |
111.41 |
60.98 (1.60) |
1.096 (0.960) |
111.45
|
61.46 (60.40) |
|
H27 |
C21 |
N19 |
C17 |
- |
- |
- |
1.091 (0.960) |
110.73 (109.45) |
59.06 (53.188) |
|
H28 |
C21 |
N19 |
C17 |
- |
- |
- |
1.090 (0.960) |
107.97 (109.48) |
178.80 (173.89) |
|
H29 |
C21 |
N19 |
C17 |
- |
- |
- |
1.092 (0.960) |
111.07 (109.46) |
61.19 (66.17) |
-: Not applicable
a: Value in braces corresponds to experiment (Ref. 17)
Table 2. Bifurcated intramolecular hydrogen bond geometry (bond lengths in Å, bond angles in degrees) for HMHC and HMNHC.
|
Molecule |
D─H•••A |
D─Ha |
H•••Aa |
D•••Aa |
D─H•••Aa |
|
HMHC |
O10─H11•••N14 |
0.976 (0.820) |
1.844 (1.963) |
2.704 (2.680) |
145.37 (145.45) |
|
|
N19─H20•••N14 |
1.007 (0.860) |
2.373 (2.358) |
2.714 (2.691) |
98.65 (103.40) |
|
HMNHC |
O10─H11•••N14 |
0.976 (0.819) |
1.848 (1.998) |
2.710 (2.711) |
145.63 (145.11) |
|
|
N19─H20•••N14 |
1.008 (0.860) |
2.301 (2.330) |
2.721 (2.699) |
103.57 (106.19) |
a: Value in braces corresponds to the experiment (Ref. 17)
Comparison of these values with corresponding X-ray values, for both HMHC and HMNHC showed good agreement (see Table 1). For example, for HMHC, as per the present computations, average bond length for mequinol intra-ring C-C bond is 1.399 Å; the average value of C-H bond length associated with the mequinol unit is 1.082 Å; and mequinol C-OH, C2-C12 and C-OCH3 bonds measure 1.354 Å, 1.450 Å, and 1.367 Å, respectively. The agreement with their corresponding X-ray values[17] at 1.384 Å, 0.93 Å, 1.36 Å, 1.450 Å, and 1.373 Å is good except for C-H bond length as expected (electron cloud around each hydrogen atom is not dense enough to scatter the X-rays effectively, as it arises from a single electron associated with an hydrogen atom). Further, existence of OH moiety and methoxy group in mequinol ring of both the molecules are likely to influence the values of bond angles ∠C5C6C1, ∠C6C1C2, and ∠C1C2C3, along with those for ∠C2C3C4, ∠C3C4C5 and ∠C4C5C6. Their corresponding calculated values at 120.71° (120.71°), 119.37° (119.46°), 119.20° (119.11°); and 120.82° (120.85°), 119.23° (119.25°), and 120.66° (120.61°) for HMHC (value in braces corresponds to HMNHC), exhibit good agreement with their corresponding X-ray values [17], near 120.05° (120.68°), 119.68° (119.69°), 119.42° (118.80°); and 120.50° (120.76°), 119.15° (119.41°), 121.17° (120.64°) for both the molecules. Our DFT computations recognised the existence of intra-molecular, hydrogen bond between O10•••H9, and O22•••H8, along with the presence of bifurcated intra-molecular hydrogen bond involving H11, H20 atoms with N14 atom (O10-H11•••N14 and N19-H20•••N14) in both the molecules HMHC and HMNHC. Corresponding parameters related to intra-molecular and bifurcated intra-molecular hydrogen bonds for both the molecules investigated, agree well with their X-ray values, as shown in Table 2.
3.2. Non linear optical (NLO) properties
When an electromagnetic radiation interacts with a NLO material, there is a change in the propagation parameters like frequency, amplitude, and phase, creating new fields [34,35]. If these changes are note-worthy, then NLO material could be useful for nonlinear optical applications such as optical logic, optical memory, optical switching and frequency shifting [36, 37]. NLO utility of a given compound is decided by the value of its first- order hyper polarizability. Therefore, density functional theory has been commonly used to explore organic NLO materials [38-41]. Table. 3 consists of all the values of the NLO parameters for both the molecules HMHC and HMNHC.
It is a normal practice to compare total dipole moment (μt) and mean first-order hyperpolarizability (βt) of a new compound with those of Urea, which are 1.3732 Debye and 372.8×10-33 cm5/esu, respectively, to examine NLO behaviour. For HMHC, the values of μt and βt are: 1.684 Debye and 1114.785×10-33 cm5/esu, respectively, whereas the corresponding values are 1.395 Debye and 1071.073×10-33 cm5/esu, for HMNHC. Thus, µt and βt for HMHC are 1.22 and 2.99 times higher in comparison with that of Urea. In the case of HMNHC, µt and βt are 1.01 and 2.87 times greater than the corresponding figures for Urea. Hence, HMHC and HMNHC are most suitable for NLO applications. It should be noted that the nonlinear optical activity originates from intra molecular charge transfer (ICT), which is connected to electron association via the π - conjugated network of electrons [42]. Hyper polarizability is helpful to understand the charge delocalization in the molecule. Maximum charge delocalization takes place along βxxy direction in HMHC and HMNHC.
Table 3. Values of dipole moment, µt(in Debye); polarizability, αt in 1.4818 × 10-25 cm3; and first order hyperpolarizability, βt (in 8.641 × 10-33cm5/e.s.u) of HMHC and HMNHC molecules.
|
Type of component |
Value with B3LYP/6-311++G(d,p) |
|
|
HMHC |
HMNHC |
|
|
µx |
-1.644 |
1.382 |
|
µy |
-0.292 |
-0.028 |
|
µz |
0.216 |
0.192 |
|
µt |
1.684 |
1.395 |
|
axx |
326.425 |
337.070 |
|
axy |
-18.608 |
19.862 |
|
ayy |
174.093 |
191.870 |
|
axz |
-1.112 |
1.569 |
|
ayz |
2.114 |
1.377 |
|
azz |
91.932 |
101.809 |
|
αt |
197.483 |
210.250 |
|
∆α |
206.085 |
205.599 |
|
βxxx |
-1100.922 |
1012.072 |
|
βxxy |
500.456 |
479.616 |
|
βxyy |
-59.382 |
154.603 |
|
βyyy |
-184.985 |
-200.774 |
|
βxxz |
65.766 |
51.658 |
|
βxyz |
-18.053 |
33.142 |
|
βyyz |
-11.254 |
-4.139 |
|
βxzz |
77.117 |
-115.535 |
|
βyzz |
-58.234 |
-78.834 |
|
βzzz |
2.771 |
0.419 |
|
βt |
1114.785 |
1071.073 |
3.3. Frontier molecular orbitals (FMO)
FMOs i.e. HOMO (highest occupied molecular orbital) considered as electron donor and LUMO (lowest unoccupied molecular orbital) treated as an electron acceptor are known for their vital role in quantum chemistry for proper understanding and correct interpretation of various electronic and electrical parameters, UV-Visible absorption bands, Chemical reactivity and charge transfer at molecular level [28, 42, 43-50].
3.3.1. UV-Visible spectra
Usefulness of UV-Visible absorption spectra is popular for quantitative analysis of organic solutions. But the process is beset by unavoidable presence of impurities in an organic synthesis, which takes place in the solution phase. There are several methods to address this problem:
- Segregation of different impurities in the solution,
- Synthesizing the impurities individually, and
- Simulating UV-Visible spectra theoretically.
The first two methods are not only financially taxing but also need considerable time. This paved the way for development of theoretical methods, that strived to achieve the common goal of simulating UV-Visible spectra. A succinct review of these methods along with DFT was made by Parrish et al [51], while presenting their own method involving extension of variational quantum eigensolver approach for calculating electronic transitions. As time-dependent density functional theory (TD-DFT) is known to provide UV-Visible spectra with acceptable accuracy [28, 52], we employed the same in the present calculations. Another compelling reason for our choice is that it conforms to the overall aim of these investigations.
Simulation of UV-Visible spectra is a three-step process [53, 54]. These steps are:
- Generation of ground state geometry,
- Analytical computation of vibrational wavenumbers in order to ensure the absence of imaginary frequencies, a mandatory condition, to ascertain that the structure correlates with a local minimum, and
- Calculation of vertical transitions to valence excited states.
The first two steps are implemented with the help of DFT, whereas in the final step TD-DFT is employed.
Quantum chemical methods outlined above provide us with a line spectrum for the UV-Visible transitions, whereas its experimental counterpart appears with a broad shape caused by line-broadening effects (natural line width, Doppler effect, Pressure broadening and thermal excitement), that are inherent to a chemical system of present kind. This underlines the need for modifying each computed line to assume a Gaussian shape [55, 56]. To this end, we adopted an integrated approach due to Bremond et al [57], to calculate UV-Visible transitions, using electronic transitions generated by TD-DFT as input. This process broadens each line spectrum into a Gaussian function by making independent optimization using a convenient value for full width at half maximum (FWHM). This approach results in an improved concurrence of recorded spectra with their corresponding computed spectra [37]. However, it should be kept in mind that perfect agreement between the observed and simulated UV-Visible spectra is impossible to achieve, as it is beyond the scope of theoretical methods currently available to account for line-broadening effects in their totality.
Simulated UV-Visible spectral parameters obtained in a solution of Dimethyl-formamide (DMF) for HMHC and HMNHC, according to the quantum chemical methods outlined in the preceding paragraphs (see section 2, to account for solvent effects), are shown in Figures 2 (a) and 2(b), respectively. They are compared with their experimental counterparts taken from reference 17 in Table 4.
Table 4. Experimental and theoretical (DFT/B3LYP/6-311++G(d,p) formalism) electronic absorption parameters for HMHC and HMNHC molecules.
|
Compound |
Absorption Maximum λmax (nm) |
Oscillator Strengths (f) |
Majorcontribution (≥10%) |
Transition |
|
|
Expta. |
Cal. |
||||
|
HMHC
|
329 |
332.02 |
0.3885 |
H→L (97%) |
n→π* |
|
260 |
263.98 |
0.4926 |
H-1→L (41%), H-2→L (55%) |
π→π* |
|
|
HMNHC
|
337 |
340.87 |
0.4075 |
H→L (97%) |
n→π* |
|
270 |
273.45 |
0.4771 |
H-1→L (92%) |
π→π* |
|
H: HOMO; L: LUMO
a: from reference [17]
TD-DFT/B3LYP//6-311++G(d,p) computations predict two electronic transitions at λmax=332 and 264 nm, with their oscillator strengths f=0.3885 and 0. 4926, for HMHC. These are in good agreement with experimental electronic transitions at λmax= 329 and 260 nm, reported by Subhashree et al. [17], for HMHC. In the same way, for HMNHC, on the basis of computations we expect two electronic absorption bands near λmax = 341 and 273 nm, with corresponding oscillator strengths f = 0.4075 and 0.4771, respectively. These values also agree well with their experimental counterparts around λmax = 337 and 270 nm. TD-DFT computations for HMHC confirm that the first of the above-mentioned experimental band originates mainly from H→L (97%) transition, identifiable with n→π* transition, whereas the latter band has its origin in the transitions that start from (H-1) and (H-2) and end on (L), i.e., (H-1) →L (41%) and (H-2) →L (55%), atributable to π→π* transition (see table 4). Similar observations can be made with regard to HMNHC from the data available in Table 4.
3.3.2. Chemical reactivity
Table 5 contains the frontier molecular orbital parameters calculated using the DFT/B3LYP/6-311++G(d, p) method, for HMHC and HMNHC, using formulae given in section 2.
The energy gap (Eg), is computed near 1.998, and 2.036 eV, for HMHC and HMNHC, respectively, which is small. Small value of energy gap is characteristic of conjugated systems. This implies high chemical reactivity as it is energetically easy to transfer electrons to high-lying LUMO by exciting electrons from low-lying HOMO. This causes accumulation of electrons in LUMO, with subsequent polarization of the molecules. Hence, a molecule with a small frontier orbital gap is easily polarizable, usually exhibits high chemical reactivity and low kinetic stability [58-60]. Both the molecules HMHC and HMNHC are stable under normal conditions, because they have negative chemical potential (µ) [61].
Table 5. Frontier molecular orbital parameters of HMHC and HMNHC molecules
|
Frontier molecular orbital parameter |
Value (eV) |
|
|
HMHC (This work) |
HMNHC (This work) |
|
|
HOMO energy |
-7.537 |
-7.560 |
|
LUMO energy |
-5.539 |
-5.524 |
|
Frontier molecular orbital energy gap |
1.998 |
2.036 |
|
Ionization energy (I) |
7.537 |
7.560 |
|
Electron affinity (A) |
5.539 |
5.524 |
|
Global hardness (η) |
0.999 |
1.018 |
|
Global softness (S) |
0.500 |
0.491 |
|
Chemical potential (µ) |
-6.538 |
-6.542 |
|
Electronegativity (χ) |
6.538 |
6.542 |
|
Global electrophilicity power (ω) |
21.373 |
21.020 |
3.4 Natural Bond Orbital (NBO) properties
NBO investigations identify inherent Lewis structure of the wave function (ψ), characterize conjugative charge transfer, and indicate the existence of inter- and intra-molecular hydrogen bonds, and show interactions between the bonds. In order to provide a convincing explanation for the above mentioned chemical processes, the NBO orbitals are selected in such a way that they have the most significant feasible amount of electron density (ED).
The stabilizing energy E(2) may be obtained from the expression:

In the above expression, is the population of the donor orbital, and are the energies of the ith and jth orbitals, respectively, and is the Fock matrix element in the NBO basis. Strong interaction between electron donors and acceptors, which envelops the structure in conjugation, implies significant value of stabilizing energy , that binds the entire structure in conjugation. Required computations are made with the help of NBO programme version 3.1 [32], which is a part of the Gaussian 09 software suit at the DFT/B3LYP/6-311++G(d,p) level of theory.
Table 6. Second-order perturbation theory analysis of FOCK matrix in NBO basis corresponding to the intra-molecular bonds of HMHC by DFT/B3LYP/6-311++G(d,p) method.
|
NBO(i) |
Type |
ED/e |
NBO(j) |
Type |
ED/e |
E(2)a (kcal/mol) |
E(j)-E(i)b (a.u) |
F(i,j)c (a.u) |
|
C1-C2 |
π |
1.61434 |
C3-C4 |
π* |
0.36844 |
19.24 |
0.29 |
0.066 |
|
C5-C6 |
π* |
0.30655 |
16.26 |
0.29 |
0.063 |
|||
|
C12-N14 |
π* |
0.25821 |
22.80 |
0.24 |
0.069 |
|||
|
π |
1.70950 |
C1-C2 |
π* |
0.44157 |
16.96 |
0.29 |
0.065 |
|
|
C5-C6 |
π* |
0.30655 |
17.73 |
0.30 |
0.065 |
|||
|
C5-C6 |
π |
1.71703 |
C1-C2 |
π* |
0.44157 |
20.79 |
0.28 |
0.071 |
|
C3-C4 |
π* |
0.36844 |
18.40 |
0.28 |
0.066 |
|||
|
LP(2)O10 |
- |
1.84408 |
C1-C2 |
π* |
0.44157 |
31.48 |
0.34 |
0.099 |
|
LP(1)N14 |
- |
1.89533 |
O10-H11 |
σ* |
0.04076 |
13.72 |
0.85 |
0.098 |
|
LP(1)N15 |
- |
1.66097 |
C12-N14 |
π* |
0.00909 |
28.36 |
0.27 |
0.079 |
|
- |
C17-S18 |
σ* |
0.47684 |
53.01 |
0.24 |
0.105 |
||
|
LP(2)S18 |
- |
1.87812 |
N15-C17 |
σ* |
0.05876 |
11.45 |
0.62 |
0.076 |
|
- |
C17-N19 |
σ* |
0.05040 |
11.70 |
0.65 |
0.079 |
||
|
LP(1)N19 |
- |
1.72744 |
C17-S18 |
σ* |
0.47684 |
62.08 |
0.23 |
0.113 |
|
LP(2)O22 |
- |
1.85128 |
C3-C4 |
π* |
0.36844 |
29.21 |
0.34 |
0.094 |
a: E(2) means energy of hyper conjugative interaction (stabilization energy).
b: Energy difference between donor (i) and acceptor (j) NBO orbitals.
c: F(i,j) is the Fock matrix element between i and j NBO orbitals.
Table 7. Second-order perturbation theory analysis of FOCK matrix in NBO basis corresponding to the intra-molecular bonds of HMNHC by DFT/B3LYP/6-311++G(d,p) method.
|
NBO(i) |
Type |
ED/e |
NBO(j) |
Type |
ED/e |
E(2)a (kcal/mol) |
E(j)-E(i)b (a.u) |
F(i,j)c (a.u) |
|
C1-C2 |
π |
1.61513 |
C3-C4 |
π* |
0.36986 |
19.24 |
0.28 |
0.066 |
|
C5-C6 |
π* |
0.30928 |
16.45 |
0.29 |
0.063 |
|||
|
C12-N14 |
π* |
0.26092 |
22.49 |
0.24 |
0.068 |
|||
|
π |
1.70927 |
C1-C2 |
π* |
0.44124 |
17.00 |
0.29 |
0.065 |
|
|
C5-C6 |
π* |
0.30928 |
17.69 |
0.30 |
0.065 |
|||
|
C5-C6 |
π |
1.71742 |
C1-C2 |
π* |
0.44124 |
20.69 |
0.28 |
0.071 |
|
C3-C4 |
π* |
0.36986 |
18.47 |
0.28 |
0.066 |
|||
|
LP(2)O10 |
- |
1.84591 |
C1-C2 |
π* |
0.44124 |
31.13 |
0.34 |
0.099 |
|
LP(1)N14 |
- |
1.89342 |
O10-H11 |
σ* |
0.04066 |
13.48 |
0.85 |
0.097 |
|
LP(1)N15 |
- |
1.66792 |
C12-N14 |
π* |
0.26092 |
29.58 |
0.26 |
0.080 |
|
LP(2)S18 |
- |
1.84591 |
C17-S18 |
σ* |
0.50675 |
58.71 |
0.23 |
0.108 |
|
- |
N15-C17 |
σ* |
0.05692 |
11.33 |
0.61 |
0.075 |
||
|
- |
C17-N19 |
σ* |
0.05695 |
12.03 |
0.67 |
0.081 |
||
|
LP(1)N19 |
- |
1.67563 |
C17-S18 |
σ* |
0.50675 |
80.32 |
0.20 |
0.120 |
|
LP(2)O25 |
- |
1.85166 |
C3-C4 |
σ* |
0.36986 |
29.12 |
0.34 |
0.094 |
a: E(2) means energy of hyper conjugative interaction (stabilization energy).
b: Energy difference between donor (i) and acceptor (j) NBO orbitals.
c: F(i,j) is the Fock matrix element between i and j NBO orbitals.
The results of such analysis [28, 62,63] for both the molecules HMHC and HMNHC are displayed in Tables 6 and 7, respectively. They consist of only major contributions of energy values (contributions below 10% are not included).
Donor-acceptor interactions may be stabilized by the delocalization of electron density between empty non-bonding or Rydberg (non-Lewis NBO) orbitals and lone pair or bonding (i.e., Lewis-type) NBO orbitals. Intra-molecular hyper conjugative interactions occur when a bonding π-orbital and an anti-bonding π*-orbital overlap. These interactions result in intra-molecular charge transfer (ICT), which stabilizes the system. As a result of these interactions, there is an increase of ED in mequinol C1-C2 and C5-C6 anti-bonding orbitals. This is reflected in the reduction of strength of corresponding bonds. The ED, for HMHC near the conjugated π-bonds of the mequinol ring is between 1.6143 and 1.7095, according to the values available in Table 7. Similarly, on the basis of values in the same Table, the corresponding values for π*-anti-bonds for HMHC are in the range 0.2582-0.4415. This causes intense charge delocalization in the molecule generating total stabilization energy 170.07 kcal mol-1 from mequinol ring alone. According to NBO analysis, the interactions π (C1-C2)→ π* (C3-C4, C5-C6 and C12-N14); π (C5-C6) → π* (C1-C2, C3-C4); LP(2)O10 → π* (C1-C2); LP(2)O22→ π*(C3-C4) exhibit high stabilization energies in the range 16.26 – 31.48 kcal mol-1.
Using the data presented in Table 7, similar results can be arrived at for HMNHC.
3.5 Molecular electrostatic Potential
Molecular electrostatic Potential (MESP) is a well established method for understanding molecular reactivity sites [64, 65]. The function of MESP also includes understanding the nature of biological recognition mechanisms. We know that, with the help of diffraction experiments [66], MESP can be determined experimentally for a given molecule just like any other real physical quantity. This is an essential attribute of MESP that sets it apart from other reactivity indices like atomic charges, which are just conceptual entities and hence have no direct relation to the physical world. MESP maps generated using the procedure stated to section 2 are available in Figure 3.
To identify the areas of the molecule with changing electron density, colour grading [67] technique is used. The colour scheme suggests that red denotes electron-rich region, blue indicates electron deficient area, Orange (light blue) colour is associated with slightly electron deficient region, yellow indicates a somewhat electron-rich region, and green indicates a neutral area [67]. Therefore, the colour grading from most negative region to the most positive area follows the order red˃yelow˃green˃orange˃blue.
It is evident from Fig.3 that the electron-rich area in HMHC and HMNHC is around oxygen atoms of the hydroxy and methoxy groups. Therefore oxygen atom is the most reactive site in mequinol ring. This is supported by the fact that these oxygen atoms participate in the formation of inter-molecular hydrogen bonds in both molecules. The sulfur atom, which is seen in figure as yellow region, is the next region in both the molecules that is prone to chemical attack by approaching species.
Three nitrogen atoms of the two molecules are places of negative potential whereas the hydrogen atoms correspond to areas of positive potential.
3.6 Fukui functions analyses
The Fukui functions provide quantitative data on the electrophilicity and nucleophilicity of each atom based on population of Mulliken charges. The electron density is a variable that is determined by the quantity of electrons present at a specific place. The electrons inside the molecule are added or subtracted in order to calculate the Fukui functions. In biological research, the local softness characteristics may be used to examine the protein-ligand bonding. [44,45, 68]. Tables 8 and 9 displays each atom of both the molecules HMHC and HMNHC along with the full Fukui functions and dual descriptor values. The order of positive calculated dual descriptor values for each atom in the case of HMHC,
N15>S18>H26>C4>O10>C17>C6>C23>H24>C1>C3>H11>H25>O22. The atom C12 has an electrophilic attack value of -0.102, it is the most detrimental site. The order accepting active sites can be organized as supervene:
C12>N14>C5>H13>C2>N19>H7>C21>H9>H8>H21>H16. For the second molecule that is HMNHC, the order of positive calculated dual descriptor values for each atom as follows,
N15>S18>O22>C4>O10>C17>C6>H27>H19>C1>H11>H20>C3>H23>H28>C21. The most determinantal site atom C12 and the value of its electrophilic attack is -0.106. The order of negative values of f(r) is C12>N14>C5>H13>C2>H7>H9>H8>N19>H29>H22>C23>H16.
Table 8. Condensed Fukui function, dual descriptor, and local softness of HMHC
|
Atom |
Mulliken atomic charges |
Fukui functions |
dual desciptors |
local softness |
||||||
|
0, 1 (N) |
N +1 (-1, 2) |
N-1 (1,2) |
fr+ |
fr- |
fr0 |
Df |
sr+ ƒr+ |
sr-ƒr- |
sr0 ƒr0 |
|
|
C1 |
-0.2794 |
-0.329737 |
-0.22104 |
-0.050 |
-0.058 |
-0.054 |
0.008 |
-0.011 |
-0.013 |
-0.012 |
|
C2 |
1.133 |
1.038187 |
1.209101 |
-0.095 |
-0.076 |
-0.085 |
-0.019 |
-0.021 |
-0.017 |
-0.019 |
|
C3 |
-0.2669 |
-0.301523 |
-0.22561 |
-0.035 |
-0.041 |
-0.038 |
0.007 |
-0.008 |
-0.009 |
-0.008 |
|
C4 |
-0.8491 |
-0.843732 |
-0.81838 |
0.005 |
-0.031 |
-0.013 |
0.036 |
0.001 |
-0.007 |
-0.003 |
|
C5 |
-0.0592 |
-0.090971 |
-0.07831 |
-0.032 |
0.019 |
-0.006 |
-0.051 |
-0.007 |
0.004 |
-0.001 |
|
C6 |
-0.2056 |
-0.166888 |
-0.22644 |
0.039 |
0.021 |
0.030 |
0.018 |
0.008 |
0.005 |
0.006 |
|
H7 |
0.10085 |
0.053236 |
0.139155 |
-0.048 |
-0.038 |
-0.043 |
-0.009 |
-0.010 |
-0.008 |
-0.009 |
|
H8 |
0.2026 |
0.140837 |
0.258051 |
-0.062 |
-0.055 |
-0.059 |
-0.006 |
-0.013 |
-0.012 |
-0.013 |
|
H9 |
0.19114 |
0.133537 |
0.2413 |
-0.058 |
-0.050 |
-0.054 |
-0.007 |
-0.013 |
-0.011 |
-0.012 |
|
O10 |
-0.2692 |
-0.319279 |
-0.1851 |
-0.050 |
-0.084 |
-0.067 |
0.034 |
-0.011 |
-0.018 |
-0.015 |
|
H11 |
0.37728 |
0.373655 |
0.386424 |
-0.004 |
-0.009 |
-0.006 |
0.006 |
-0.001 |
-0.002 |
-0.001 |
|
C12 |
-0.3731 |
-0.467459 |
-0.38078 |
-0.094 |
0.008 |
-0.043 |
-0.102 |
-0.021 |
0.002 |
-0.009 |
|
H13 |
0.18271 |
0.111502 |
0.22199 |
-0.071 |
-0.039 |
-0.055 |
-0.032 |
-0.016 |
-0.009 |
-0.012 |
|
N14 |
0.00156 |
-0.112405 |
0.032239 |
-0.114 |
-0.031 |
-0.072 |
-0.083 |
-0.025 |
-0.007 |
-0.016 |
|
N15 |
0.10665 |
0.158985 |
0.137639 |
0.052 |
-0.031 |
0.011 |
0.083 |
0.011 |
-0.007 |
0.002 |
|
H16 |
0.22912 |
0.189534 |
0.267069 |
-0.040 |
-0.038 |
-0.039 |
-0.002 |
-0.009 |
-0.008 |
-0.008 |
|
C17 |
0.05818 |
0.108845 |
0.038507 |
0.051 |
0.020 |
0.035 |
0.031 |
0.011 |
0.004 |
0.008 |
|
S18 |
-0.4757 |
-0.711199 |
-0.19095 |
-0.236 |
-0.285 |
-0.260 |
0.049 |
-0.051 |
-0.062 |
-0.057 |
|
N19 |
-0.3716 |
-0.406065 |
-0.35429 |
-0.035 |
-0.017 |
-0.026 |
-0.017 |
-0.008 |
-0.004 |
-0.006 |
|
H20 |
0.2711 |
0.263455 |
0.283876 |
-0.008 |
-0.013 |
-0.010 |
0.005 |
-0.002 |
-0.003 |
-0.002 |
|
H21 |
0.30429 |
0.264372 |
0.336305 |
-0.040 |
-0.032 |
-0.036 |
-0.008 |
-0.009 |
-0.007 |
-0.008 |
|
O22 |
-0.1649 |
-0.185372 |
-0.10255 |
-0.020 |
-0.062 |
-0.041 |
0.042 |
-0.004 |
-0.014 |
-0.009 |
|
C23 |
-0.3404 |
-0.332227 |
-0.35415 |
0.008 |
0.014 |
0.011 |
-0.006 |
0.002 |
0.003 |
0.002 |
|
H24 |
0.1563 |
0.140348 |
0.182353 |
-0.016 |
-0.026 |
-0.021 |
0.010 |
-0.003 |
-0.006 |
-0.005 |
|
H25 |
0.18327 |
0.148578 |
0.220914 |
-0.035 |
-0.038 |
-0.036 |
0.003 |
-0.008 |
-0.008 |
-0.008 |
|
H26 |
0.15695 |
0.141785 |
0.182677 |
-0.015 |
-0.026 |
-0.020 |
0.011 |
-0.003 |
-0.006 |
-0.004 |
Table 9. Condensed Fukui function, dual descriptor, and local softness of HMNHC
|
Atom |
Mulliken atomic charges |
Fukui functions |
dual descriptors |
local softness |
||||||
|
0, 1 (N) |
N +1 (-1, 2) |
N-1 (1,2) |
fr+ |
fr- |
fr0 |
Df |
sr+ ƒr+ |
sr-ƒr- |
sr0 ƒr0 |
|
|
C1 |
-0.3051 |
-0.35454 |
-0.24852 |
-0.049 |
-0.057 |
-0.053 |
0.007 |
-0.011 |
-0.012 |
-0.012 |
|
C2 |
1.16505 |
1.080102 |
1.235264 |
-0.085 |
-0.070 |
-0.078 |
-0.015 |
-0.019 |
-0.015 |
-0.017 |
|
C3 |
-0.2242 |
-0.261929 |
-0.18208 |
-0.038 |
-0.042 |
-0.040 |
0.004 |
-0.008 |
-0.009 |
-0.009 |
|
C4 |
-0.8323 |
-0.827185 |
-0.80118 |
0.005 |
-0.031 |
-0.013 |
0.036 |
0.001 |
-0.007 |
-0.003 |
|
C5 |
-0.0713 |
-0.102761 |
-0.09025 |
-0.031 |
0.019 |
-0.006 |
-0.050 |
-0.007 |
0.004 |
-0.001 |
|
C6 |
-0.2233 |
-0.185188 |
-0.24486 |
0.038 |
0.022 |
0.030 |
0.016 |
0.008 |
0.005 |
0.007 |
|
H7 |
0.09141 |
0.044515 |
0.128624 |
-0.047 |
-0.037 |
-0.042 |
-0.010 |
-0.010 |
-0.008 |
-0.009 |
|
H8 |
0.20153 |
0.139886 |
0.256635 |
-0.062 |
-0.055 |
-0.058 |
-0.007 |
-0.013 |
-0.012 |
-0.013 |
|
H9 |
0.18849 |
0.130876 |
0.238108 |
-0.058 |
-0.050 |
-0.054 |
-0.008 |
-0.013 |
-0.011 |
-0.012 |
|
O10 |
-0.267 |
-0.316248 |
-0.18499 |
-0.049 |
-0.082 |
-0.066 |
0.033 |
-0.011 |
-0.018 |
-0.014 |
|
H11 |
0.38832 |
0.383927 |
0.397456 |
-0.004 |
-0.009 |
-0.007 |
0.005 |
-0.001 |
-0.002 |
-0.001 |
|
C12 |
-0.4114 |
-0.51383 |
-0.41502 |
-0.102 |
0.004 |
-0.049 |
-0.106 |
-0.022 |
0.001 |
-0.011 |
|
H13 |
0.17954 |
0.107545 |
0.220572 |
-0.072 |
-0.041 |
-0.057 |
-0.031 |
-0.016 |
-0.009 |
-0.012 |
|
N14 |
0.03165 |
-0.081241 |
0.06134 |
-0.113 |
-0.030 |
-0.071 |
-0.083 |
-0.025 |
-0.006 |
-0.016 |
|
N15 |
0.11243 |
0.156754 |
0.150876 |
0.044 |
-0.038 |
0.003 |
0.083 |
0.010 |
-0.008 |
0.001 |
|
H16 |
0.24408 |
0.204903 |
0.281946 |
-0.039 |
-0.038 |
-0.039 |
-0.001 |
-0.009 |
-0.008 |
-0.008 |
|
C17 |
-0.0479 |
-0.018662 |
-0.05429 |
0.029 |
0.006 |
0.018 |
0.023 |
0.006 |
0.001 |
0.004 |
|
S18 |
-0.4128 |
-0.621359 |
-0.14736 |
-0.209 |
-0.265 |
-0.237 |
0.057 |
-0.045 |
-0.058 |
-0.052 |
|
N19 |
-0.2596 |
-0.266939 |
-0.25913 |
-0.007 |
0.000 |
-0.004 |
-0.007 |
-0.002 |
0.000 |
-0.001 |
|
H20 |
0.31752 |
0.313185 |
0.326984 |
-0.004 |
-0.009 |
-0.007 |
0.005 |
-0.001 |
-0.002 |
-0.002 |
|
C21 |
-0.3296 |
-0.320018 |
-0.3392 |
0.010 |
0.010 |
0.010 |
0.000 |
0.002 |
0.002 |
0.002 |
|
O22 |
-0.1657 |
-0.18597 |
-0.10542 |
-0.020 |
-0.060 |
-0.040 |
0.040 |
-0.004 |
-0.013 |
-0.009 |
|
C23 |
-0.3386 |
-0.33033 |
-0.35209 |
0.008 |
0.013 |
0.011 |
-0.005 |
0.002 |
0.003 |
0.002 |
|
H24 |
0.15675 |
0.141427 |
0.18187 |
-0.015 |
-0.025 |
-0.020 |
0.010 |
-0.003 |
-0.005 |
-0.004 |
|
H25 |
0.18314 |
0.148458 |
0.2202 |
-0.035 |
-0.037 |
-0.036 |
0.002 |
-0.008 |
-0.008 |
-0.008 |
|
H26 |
0.15623 |
0.140201 |
0.181598 |
-0.016 |
-0.025 |
-0.021 |
0.009 |
-0.003 |
-0.006 |
-0.005 |
|
H27 |
0.17755 |
0.15163 |
0.198372 |
-0.026 |
-0.021 |
-0.023 |
-0.005 |
-0.006 |
-0.005 |
-0.005 |
|
H28 |
0.11707 |
0.092585 |
0.144781 |
-0.024 |
-0.028 |
-0.026 |
0.003 |
-0.005 |
-0.006 |
-0.006 |
|
H29 |
0.17797 |
0.150206 |
0.199768 |
-0.028 |
-0.022 |
-0.025 |
-0.006 |
-0.006 |
-0.005 |
-0.005 |
Geometry optimization made for HMHC and HMNHC using DFT/B3LYP/6-311 + + G(d,p) method showed that the structure parameters so obtained were in reasonably, in good agreement not only with their corresponding X-ray values for both the molecules, but also with their corresponding counterparts in related molecules. The calculations indicated the existence of bifurcated intra-molecular hydrogen bond between (H11, H20) atoms and N14 atom (O10-H11•••N14 and N19-H20•••N14), in addition to the presence of intra-molecular hydrogen bond between O10•••H9 and O22•••H8 in the investigated molecules. Simulated electronic transitions, two each for HMHC and HMNHC, obtained employing Time-Dependent variant of DFT using above mentioned exchange correlation functional and basis set in a solution of dimethylformamide, along with IEF-PCM model, exhibited excellent coincidence with their corresponding experimental counterparts. This observation also concurs with that made for related systems. FMO analysis involving HOMO and LUMO orbitals led to the conclusion that their origin was in n→π* and π→π* transitions, which agree with that found for related compounds. Further, the value of energy gap (∆E), as provided by HOMO and LUMO values, being relatively small around 2 eV, which is also true for related molecules, suggested that both HMHC and HMNHC were highly reactive. Moreover, chemical potential (µ), being negative for both the molecules, emphasized that they were stable under normal conditions, as in the case of related systems. Comparison of NLO parameters µt and βt with those of Urea, revealed that both the molecules could be employed gainfully for NLO applications. It is interesting to note that this is also true of related molecules. A deeper insight into NLO behavior of these molecules in terms of ICT was provided by analysing hyperconjugative interactions using NBO analysis. MESP analysis helped to locate most reactive sites in both the molecules, which were around the oxygen atoms of the hydroxyl and the methoxy groups. By using Fukui functions, we have determined the reactivity of a molecule to nucleophilic and electrophilic attacks. Thus, Fukui functions have proven to be valuable tools in predicting chemical reactivity. In the molecule HMHC, most positive atom is N15 and the most negative site C12, whereas in the case of HMNHC, the highest positive atom site is N15 and the highest negative atom is C12.
Acknowledgments
The first author (KSS) is grateful to the management of SR University, Warangal, India, for permitting him to undertake the reported investigations. The second author (LR) thanks the management of Malla Reddy Engineering College(A), Secundrabad-500100, Telangana, India for permitting him to undertake the reported investigations.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding Declaration in the manuscript
There was no funding sources.
Author Contribution
K. Srishailam: Original draft preparation L. Ravindranath: ConceptualizationGaddam. Ramesh: Methodology,D. Praveena: Drafting figuresSunil kumar V: Drafting tables Danikonda. Suresh Kumar: Investigation, and validationS. Muthu: Reviewing and editing manuscript G. Ramana Rao: Reviewing and editing manuscript
- (a) Brockrnan RW, Sidweli RW Shaddix S (1970) Heterocyclic thiosemicarbazones: correlation between structure, inhibition of ribonucleotide reductase, and inhibition of DNA viruses. Proc Soc Exp Biol Med 133:609-614. (b) Park KC, Fouani L, Jansson PJ, Wooi D, Sahni S, Lane D J R , Palanimuthu D, Lok H C, Kovacevic Z, Huang MLH, Kalinowski DS, Richardson D R (2016) Copper and conquer: copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics, Metallomics. 8:874-886.
- Haribabu J, Jeyalakshmi K, Arun Y, Bhuvanesh NSP, Perumal PT, Karvembu R(2016) Synthesis of Ni (II) complexes bearing indole-based thiosemicarbazone ligands for interaction with biomolecules and some biological applications. J Biol Inorg Chem R http://dx.doi.org/10.1007/s00775- 016-1424-1
- Muralisankar M, Bhuvanesh NSP, Sreekanth A (2016) Synthesis, X-ray crystal structure, DNA/protein binding and DNA cleavage studies of novel copper (II) complexes of N-substituted isatin thiosemicarbazone ligands. New J Chem 402:661-2679.
- Yang XB, Wang Q, Huang Y, Fu PH, Zhang JS, Zeng RQ (2012) Synthesis, DNA interaction and antimicrobial activities of copper (II) complexes with Schiff base ligands derived from kaempferol and polyamines . Inorg Chem Commun 25: 55-59.
- Castineiras A, Hermida NF, Santos IG, Rodriguez L G (2012) Neutral Ni II, Pd II and Pt II ONS-pincer complexes of 5-acetylbarbituric-4 N-dimethylthiosemicarbazone: synthesis characterization and properties. Dalton Trans 41:13486-13495.
- (a) Maia PIS, Pavan FR, Leite CQF, Lemos SS, Sousa GF, Batista AA, Nascimento OR, Ellena J, Castellano EE, Niquet E, Deflon VM (2009) Vanadium complexes with thiosemicarbazones: Synthesis characterization crystal structures and anti-Mycobacterium tuberculosis activity. Polyhedron 28:398-406. (b) Yousef TA, Reash GMAE, Gammal OAE, Bedier RA (2013) Synthesis, characterization, optical band gap, in vitro antimicrobial activity and DNA cleavage studies of some metal complexes of pyridyl thiosemicarbazone. J Mol Struct 1035 : 307-317.
- (a) Aly MM, Mohamed YA, Bayouki KAME, Basyouni WM, Abbas SY (2010) Synthesis of some new 4(3H)-quinazolinone-2-carboxaldehyde thiosemicarbazones and their metal complexes and a study on their anticonvulsant analgesic cytotoxic and antimicrobial activities – Part-1. Eur J Med Chem 45:3365-3373. (b) Acharyya R, Peng SM, Lee GH, Bhattacharya S (2003) An Unprecedented Oxidative Migration of a Methyl Group from 2-(2 ', 6 '-Dimethylphenylazo)-4-methylphenol Mediated by Ruthenium and OsmiumInorg. Chem 42:7378- 7380.
- Kalaivani P, Umadevi C, Prabhakaran R, Dallemer F, Mohan PS, Natarajan K (2014) An Unprecedented Oxidative Migration of a Methyl Group from 2-(2 ', 6 '-Dimethylphenylazo)-4-methylphenol Mediated by Ruthenium and Osmium New palladium(II) complexes of 3-methoxysalicylaldehyde-4(N)-substituted thiosemicarbazones: Synthesis spectroscopy X-ray crystallography and DNA/protein binding study. Polyhedron. 80: 97-105.
- Banerjee D, Yogeeswari P, Bhat P, Thomas A, Srividya M, Sriram V (2011) Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur J Med Chem 46:106-121.
- Muralisankar M, Haribabu J, Bhuvanesh NSP, Karvembu R, Sreekanth A (2016) Synthesis X-ray crystal structure DNA/protein binding DNA cleavage and cytotoxicity studies of N(4) substituted thiosemicarbazone based copper(II)/nickel(II) complexes. Inorg Chim Acta 449:82-95.
- Ramachandran E, Senthil Raja D, Rath N P, Natarajan K (2013) Role of Substitution at Terminal Nitrogen of 2-Oxo-1,2-dihydroquinoline-3-Carbaldehyde Thiosemicarbazones on the Coordination Behavior and Structure and Biological Properties of Their Palladium(II) Complexes Inorg Chem 52:1504-1514.
- Kalaivani P, Prabhakaran R, Poornima P, Dallemer F, Vijayalakshmi K, Vijaya Padma V, Natarajan K (2012) Versatile Coordination Behaviour of Salicylaldehydethiosemicarbazone in Ruthenium(II) Carbonyl Complexes: Synthesis Spectral X-ray Electrochemistry DNA Binding Cytotoxicity and Cellular Uptake Studies Organometallics 31:8323-8332.
- Stacy A E, Palanimuthu D, Bernhardt P V, Kalinowski D S, Jansson P J, Richardson D R (2016) Zinc(II)–Thiosemicarbazone Complexes Are Localized to the Lysosomal Compartment Where They Transmetallate with Copper Ions to Induce Cytotoxicity. J Med Chem 59:4965-4984.
- Richardson D R, Kalinowski D S, Richardson V, Sharpe P C, Lovejoy D B, Islam M, Bernhardt P V (2009) 2-Acetylpyridine Thiosemicarbazones are Potent Iron Chelators and Antiproliferative Agents: Redox Activity Iron Complexation and Characterization of their Antitumor Activity.J Med Chem 52:1459-1470.
- Finch RA, Liu MC, Cory AH, Cory JG, Sartorelli AC (2000) Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity Adv Enzyme Regul 39:3-12.
- Traynor A M, Lee J W, Bayer G K, Tate J M, Thomas S P, Mazurczak M, Graham D L, Kolesar J M, Schiller J H A (2010) A phase II trial of Triapine® (NSC# 663249) and gemcitabine as second line treatment of advanced non-small cell lung cancer: Eastern Cooperative Oncology Group Study 1503. Investig New Drugs 28:91-97
- Subhashree G R , Haribabu J, Saranya S, Yuvaraj P , Anantha Krishnan D , Karvembu R, Gayathri D (2017) In vitro antioxidant, antiinflammatory and in silico molecular docking studies of thiosemicarbazones. J Mol Struct 1145:160-169.
- Srishailam K, Ramaiah , Laxma Reddy K, Venkatram Reddy B, Ramana Rao G (2021) Synthesis and evaluation of molecular structure from torsional scans study of molecular characteristics using spectroscopic and DFT methods of some thiosemicarbazones and investigation of their anticancer activity.Chemical Papers.75:3635-3647
- Ramaiah K, Srishailam K, Laxma Reddy K, Venkatram Reddy B, Ramana Rao G (2019) Synthesis crystal and molecular structure and characterization of 2- ((2- aminopyridin-3-yl)methylene)-N-ethylhydrazinecarbothioamide using spectroscopic (1H and 13C NMR, FT-IR, FT-Raman, UV-Vis) and DFT methods and evaluation of its anticancer activity. J of Mol Struct 1184: 405-417.
- Ramaiah K, Srishailam K , Laxma Reddy K, Venkatram Reddy B, Ramana Rao G (2023)Insights into synthesis structural energetic vibrational anticancer activity and molecular characteristics of 2-((2-aminopyridin-3-l)methylene)Nphenylhydrazinecarbothioamide as evaluated using spectroscopic and DFT investigations J Mol Struct 1294 :136339 .
- Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M. ... Fox D J (2009) Gaussian09 Revision E.01 Gaussian Inc Wallingford CT.
- Becke A D (1993) Density-functional thermo chemistry III The role of exact exchange J Chem Phys 98:5648-5652.
- Lee C, Yang W, Parr R G (1988) Development of the Colic-Salvetti correlation energy formula into a functional of the electron density Phys Rev B 37:785-789.
- Petersson G A, Al-Laham M A (1991) A complete basis set model chemistry II Open-shell systems and the total energies of the first-row atoms. J Chem Phys 94:6081-6090.
- Petersson G A, Bennett A, Tensfeldt T G, AI-Laham M A, Shirley W A, Mantzaris J (1988) A complete basis set model chemistry I the total energies of closed-shell atoms and hydrides of the first-row elements. J Chern Phys 89:2193-2218.
- Dennington R et al (2009) Gauss View Version 6.0 Semichem Inc Shawnee Mission.
- Scalmani G , Frisch MJ (2010) “Continuous surface charge polarizable continuum models of solvation I General formalism.” J Chem Phys 132:1-15
- Dege N, Gökce H, Doğan O. E, Alpaslan G, Ağar T, Muthu S, and Sert Y (2022) Quantum computational, spectroscopic investigations on N-(2-((2-chloro-4,5-dicyanophenyl)amino)ethyl)-4-methylbenzenesulfonamide by DFT/TD-DFT with different solvents molecular docking and drug-likeness researches. Colloids and Surfaces A: Physicochemical and Engineering Aspects 638:128311.
- Buckingham AD (1967) Permanent and induced molecular moments and long-range inter molecular forces. Adv Chem Phys 12:107-142.
- Read AEL, Curtiss LA, Weinhold F (1988) Intermolecular Interactions from a Natural bond Orbital. Donar-Acceptor Viewpoint Chem Re 88:899-926.
- Politzer P, Laurence P R, and Jayasuriya K (1985) structure-activity correlation in mechanism studies and predictive toxicology Environmental Health Perspectives 61:191-02.
- Glendening E D, Reed A E, Carpenter J E, and Weinhold F (1998) NBO Version 3.1 (University of Wisconsin,Madison: TCI.
- Politzer P, Laurence P R, and Jayasuriya K (1985) structure-activity correlation in mechanism studies and predictive toxicology. Environmental Health Perspectives 61:191-02.
- Pongor G, Fogarasi G (1985) Theoretical Prediction of Vibrational Spectra: The Out-of-Plane Force Field and Vibrational Spectra of Pyridine. J Mol Spectrosc 114:445-453.
- Sun Y X (2009) et al Experimental and density functional studies on 4-(3,4-dihydroxybenzylideneamino)antipyrine, and 4-(2,3,4-trihydroxybenzylideneamino)antipyrine. J Mol Struct Theochem 904:74-82.
- Sun Y X (2009) et al, Experimental and density functional studies on 4-(4- cyanobenzylideneamino)antipyrine. Mol Phys 107:223-235.
- Andraud C, Brotin T, Garcia C, Pelle F, Goldner P, Bigot B, Collet A (1994) Theoretical and Experimental Investigations of the Nonlinear Optical Properties of Vanillin Polyenovanillin and Bisvanillin Derivatives. J Am Chem Soc 116:2094-2102.
- Sajan D(2006) et al Structural and electronic contributions to hyperpolarizability in methyl p-hydroxy benzoate. J Mol Struct 785:43–53.
- Ahmed A B (2010) et al Crystal studies, vibrational spectra and non-linear optical properties of l-histidine chloride monohydrate Spectrochim. Acta A. 75:293-298.
- Abraham J P (2009) et al, Efficient p-electron conjugated push–pull nonlinear optical chromophore 1-(4-methoxyphenyl)-3-(3,4-dimethoxyphenyl)-2-propen-1-one: A vibrational spectral study. J Mol Struct 917: 27–36.
- Sagdinc S G, Esme A (2010) A Theoretical and vibrational studies of 4,5-diphenyl-2-2 oxazole propionic acid (oxaprozin) Spectrochim. Acta A.
- Arivazhagan M Jeyavijayan S (2011) Vibrational spectroscopic first-order hyperpolarizability and HOMO LUMO studies of 1,2-dichloro-4-nitrobenzene based on Hartree–Fock and DFT calculations Spectrochim. Acta Part A 79:376-383.
- Gökce H, Sert Y, Alpaslan G, El-Azab A S, Alanazi, M M , Al-Agamy M H M, and Abdel-Aziz A A-M (2019) “Hirshfeld Surface, Molecular Docking Study, Spectroscopic Characterization and NLO Profile of 2-Methoxy-4,6-Diphenylnicotinonitrile.” ChemistrySelect 4:9857–70. doi 10.1002/slct.201902391.
- Fukui A (1982) A study of correlation of the order of chemical reactivity of a sequence of binary compounds of nitrogen and oxygen in terms of frontier orbital theory. Science 218: 747–754.
- Par RGr, zentpaly LV, and Liu S (1999) “Electrophilicity Index” Journal of the American Chemical Society 121. no. 9:1922–1924.
- Ho Choi C, and Kertesz M (1997) “Conformational Information from Vibrational Spectra of Styrene trans-Stilbene and cis-Stilbene.” The Journal of Physical Chemistry A 101 no. 20: 3823–31.
- Pearson R G (1989) “Absolute Electronegativity and Hardness: Applications to Organic Chemistry.” The Journalof Organic Chemistry 54 no 6:1423–30.
- Parr RG, and Pearson RG (1983) “Absolute Hardness: Companion Parameter to Absolute Electronegativity .”Journal of the American Chemical Society 105 no 26:7512–6.
- Geerlings P, Proft F De, and Langenaeker W (2003) “Conceptual Density Functional Theor.” Chemical Reviews103 no 5:1793–8731873.
- Zhan CG, Nichols J A, and Dixon D A (2003) “Ionization Potential Electron Affinity Electronegativity Hardness and Electron Excitation Energy: Molecular Properties from Density Functional Theory OrbitalEnergies.” The Journal of Physical Chemistry A 107 no. 20:4184–95.
- Parrish RM, Hohenstein EG, McMahon PL, and Martınez TJ (2019) “Quantum Computation of ElectronicTransitions Using a Variational Quantum Eigensolver.” Physical Review Letters 122 no. 23 :230401.
- Jacquemin D, Perpete EA, Ciofini I, and Adamo C (2009) “Accurate Simulation of Optical Properties in Dyes.”Accounts of Chemical Research 42 no. 2: 326–34.
- Jacquemin D, and Perpete E A (2006) “Ab Initio Calculations of the Colour of Closed-Ring Diarylethenes: TD-DFT Estimates for Molecular Switches.” Chemical Physics Letters 429 no. 1–3:147–52 doi: 10.1016/j.cplett.2006.08.028.
- Preat J, Michaux C, Lewalle A, Perpete E A, and Jacquemin D (2008) “Delocalisation in Conjugated TriazeneChromophores: Insights from Theory.” Chemical Physics Letters 451 no 1–3: 37–42 doi:10.1016/j.cplett.2007.11.056.
- Maric D, and Burrows JP (1996), “Application of a Gaussian Distribution Function to Describe Molecular UV Visible Absorption Continua.1 Theory” The Journal of Physical Chemistry 100 no 21:8645–59.
- Maric D, Crowley J N, and Burrows J P (1997) “Application of a Gaussian Distribution Function to Describe Molecular UV-Visible Absorption Continua. 2 The UV Spectra of RO2 Radicals” The Journal of Physical Chemistry A 101 no. 14: 2561–7.
- Bremond EAG, Kieffer J, and Adamo C (2010) “A Reliable Method for Fitting TD-DFT Transitions to Experimental UV–Visible Spectra” Journal of Molecular Structure: Theochem 954 no 1–3:52–6. doi:10.1016/j.theochem.2010.04.038.
- Sinha L (2011) et al, Raman, FT-IR spectroscopic analysis and first-order hyperpolarisability of 3-benzoyl-5- chlorouracil by first principles. J Mol Simul 37:153-163.
- Lewis DF V (1994) et al, Inhibition of rat hepatic aryl hydrocarbon hydroxylase activity by a series of 7-hydroxy coumarins: QSAR studies Xenobiotica 24:829-838.
- Kosar B, AlbayrakC (2011) Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol Spectrochim. Acta Part A 78:160-167.
- Nakano M(2002) et al, Theoretical study on second hyperpolarizabilities of phenylacetylene dendrimer: Toward an understanding of structure-property relation in NLO responses of fractal antenna dendrimers. J Am Chem Soc 124:9648-9655.
- Kumar A, Gadre SR, Mohan N and Suresh CH (2014) Lone pairs: An Electostatic viewpoint. J Phys Chem A 118:2,526-532.
- Rahm M, Christe K O (2013), Quantifying the Nature of Lone Pair Domain. Chem Phys Chem 14:3714-3725.
- Peter Politzer, and Jane Murray S (2002) “The fundamental nature and role of the electrostatic potential in atoms and molecules.” Theoretical Chemistry Accounts 108:134-142 doi.org/10.1007/s00214-002-0363-9.
- Luque F Javier, López Josep Maria, and Modesto Orozco (2000) “Electrostatic interactions of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects.”Theoretical Chemistry Accounts103: 343-45 doi org/10.1007/s002149900013.
- Fink M, and Bonhan R A, “Electrostatic Potential of Free Molecules Derived from Electron Diffraction Results” Chemical Applications of Atomic and Molecular Potentials(1981) 93-122, doi: 10.1007/978-1-4757-9634-6_7.
- Pearson R G (1986) Absolute electronegativity and hardness correlated with molecular orbital theory Proceedings of the National Academy of Sciences of the United States of America 83: 8440-41.doi: 10.1073/pnas 83.22.8440.
- Sumithra M, Sundaraganesan N, Rajesh R, Ilangovan V, Ahmad Irfan, Muthu S (2023) Electron density, charge transfer, solvent effect and molecular spectroscopic studies on 2,2-Dimethyl-N-pyridin-4-yl-propionamide – A potential antioxidant.Comput Theor Chem 1223:114103. https://doi.org/10.1016/j.comptc.2023.114103
No competing interests reported.
{kind=link}