Patient material. FFPE or frozen tumor samples representing pediatric patient samples encountered on the typical pathology service were evaluated. The samples represented 650 samples expected to be present in the reference series and 300, true negative samples representative of non-brain solid tumors known to be absent from the reference series (Table S9).
Training and independent testing data sets
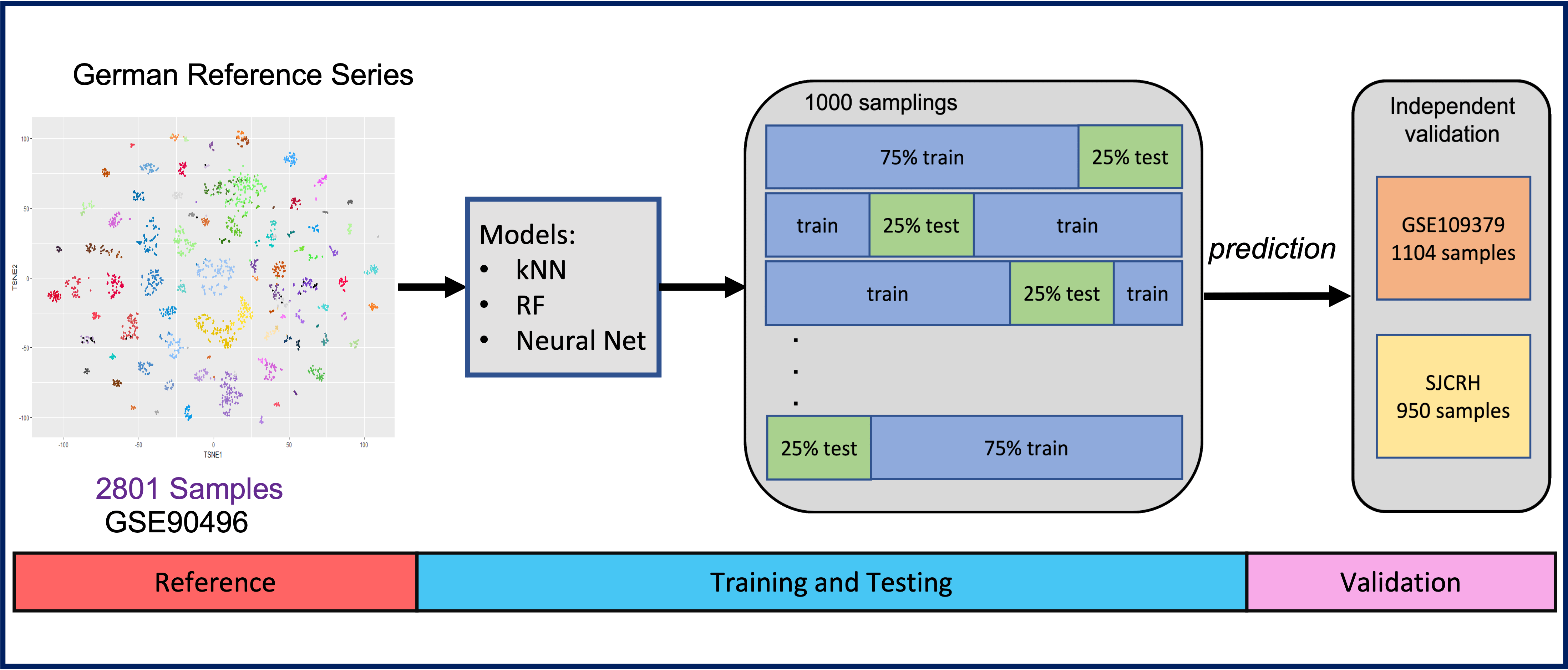
All supervised models were trained on the genome-wide DNA methylation profiles from the CNS tumor reference cohort (GSE90496), consisting of 2,801 samples from 91 methylation classes 6. All classifiers were independently validated with two methylation data sets, including the 950 CNS tumor samples from the St. Jude Children's Research Hospital (SJCRH) and 1,104 CNS tumor samples from GSE109379 6.
Data generation and methylation array processing. We analyzed the 950 independent test samples using Illumina Methylation BeadChip (EPIC) arrays according to the manufacturer's instructions. In summary, DNA was isolated from formalin-fixed paraffin-embedded (FFPE) tumor tissue using the Maxwell® Clinical Sample Concentrator system (Promega, Madison, WI). Following extraction, DNA was quantified using a Qubit fluorometer and quantitation reagents (Thermo Fisher Scientific, Waltham, MA), and bisulfite converted using the Zymo EZ DNA methylation kit (Zymo Research, Irvine, CA). The overall DNA input amount was approximately 250 ng. DNA methylation profiling was carried out with the Infinium HumanMethylationEPIC BeadChip (850K) array (Illumina Inc., San Diego, CA) on the Illumina iScan platform.
All methylation data analyses, including those from GSE90496 and GSE109379, were performed in R (http://www.r-project.org, version 3.5.3), using several packages from Bioconductor and other repositories. Specifically, array data were preprocessed using the minfi package (v.1.28.4) 34. Background correction with dye-bias normalization was performed for all samples using noob (normal-exponential out-of-band) with the "single" dye method 35. Batch effects such as hybridization time and other technical variables were removed using removeBatchEffect from the limma package (v.3.38.3) 36. Probe filtering was performed after normalization. Specifically, we removed probes located on sex chromosomes, probes containing nucleotide polymorphism (dbSNP132 Common) within five base pairs, including the targeted CpG-site, or mapping to multiple sites on hg19 (allowing for one mismatch), as well as cross-reactive probes.
Semi-supervised analysis. We developed a combination approach including a self-training with editing using a support vector machine (SETRED-SVM) as the base learner model with an L2-penalized, multinomial logistic regression model to obtain high confidence labels from a few reference instances 29. We applied this approach on GSE109379 and the SJ samples to get labels for the independent validation purpose of the supervised models. The ssc R package (v2.1-0) was used to build and train the SETRED-SMV semi-supervised model. First, the standard deviation for each probe across all 2,801 samples from GSE90496 was calculated. Input features for SSL models were the 5072 probes with a standard deviation greater than 0.3 across all 2801 samples. We used the best SETRED-SVM model to predict the methylation class for 1104 GSE109379 and 950 SJ samples. The SSL scores were calibrated with an L2-penalized, multinomial logistic regression. Scores above the 0.8 threshold were considered correctly classifiable 29.
The random forest algorithm and development. The random forest algorithm was reconstructed from Capper's algorithm 6 using the randomForest R package (v.4.6–14) 37,38. This model was trained based on the 408,862 overlapping probes of the 450K and 850K array probes. First, the 10,000 features (or probes) with the highest importance scores were selected by splitting the 408,862 intersecting probes into 43 sets of approximately 9500 probes. Next, one hundred trees were fitted for each set using 639 randomly sampled candidate features at each split (mtry = square root of 408,862). The subclass labels, stratified subsampling methods, and the number of trees in the forest were followed as in 6. This framework can produce a model that either predicts the methylation class or the methylation "family" scores 6 that represent clinically-equivalent families on which Capper et al. witnessed their best error rates. Next, a multinomial logistic regression was used to calibrate the prediction scores from all cross-validation splits as previously described 6. The family scores were then generated as the sum of all methylation class scores from the trained random forest.
The k-nearest neighbor algorithm and development (kNNmod)
An exact bootstrap k-nearest neighbor model (kNNmod) was built as described in 39. The model was trained on score vectors constructed based on the difference in median beta values of the top 100 hyper- and hypo-methylated probes. Each set of 100 top probes was selected based on the mean ß values in a methylation group and the absolute z-scores computed by taking the differences between mean beta values of two CNS methylation groups divided by the square root of the sum of the variance in each group. Hence, each methylation group had a list of 200 probes that were either most hypo- or hypermethylated based on the absolute z-scores. Each sample had a vector of scores, i.e. one score per methylation group. Each score was computed by taking the median ß values of the top 100 hypermethylated probes and subtracting that from the top 100 hypomethylated probes. Euclidean distance on these vectors was used to measure the distance between each pair of samples. The entire Euclidean distance matrix on the methylation group score vectors was computed for all pairwise samples.
To classify a new sample, kNNMod ordered all other samples by their distance from the new observation and derived the probability that those neighbors would be included among the k nearest neighbors in the binomial distribution. We used k = 5 neighbors for classification because some subgroups were very rare. For each new sample, the exact bootstrap probability of assignment to each methylation group can be conditionally computed on the training data set and the resulting probe selection and group score definition.
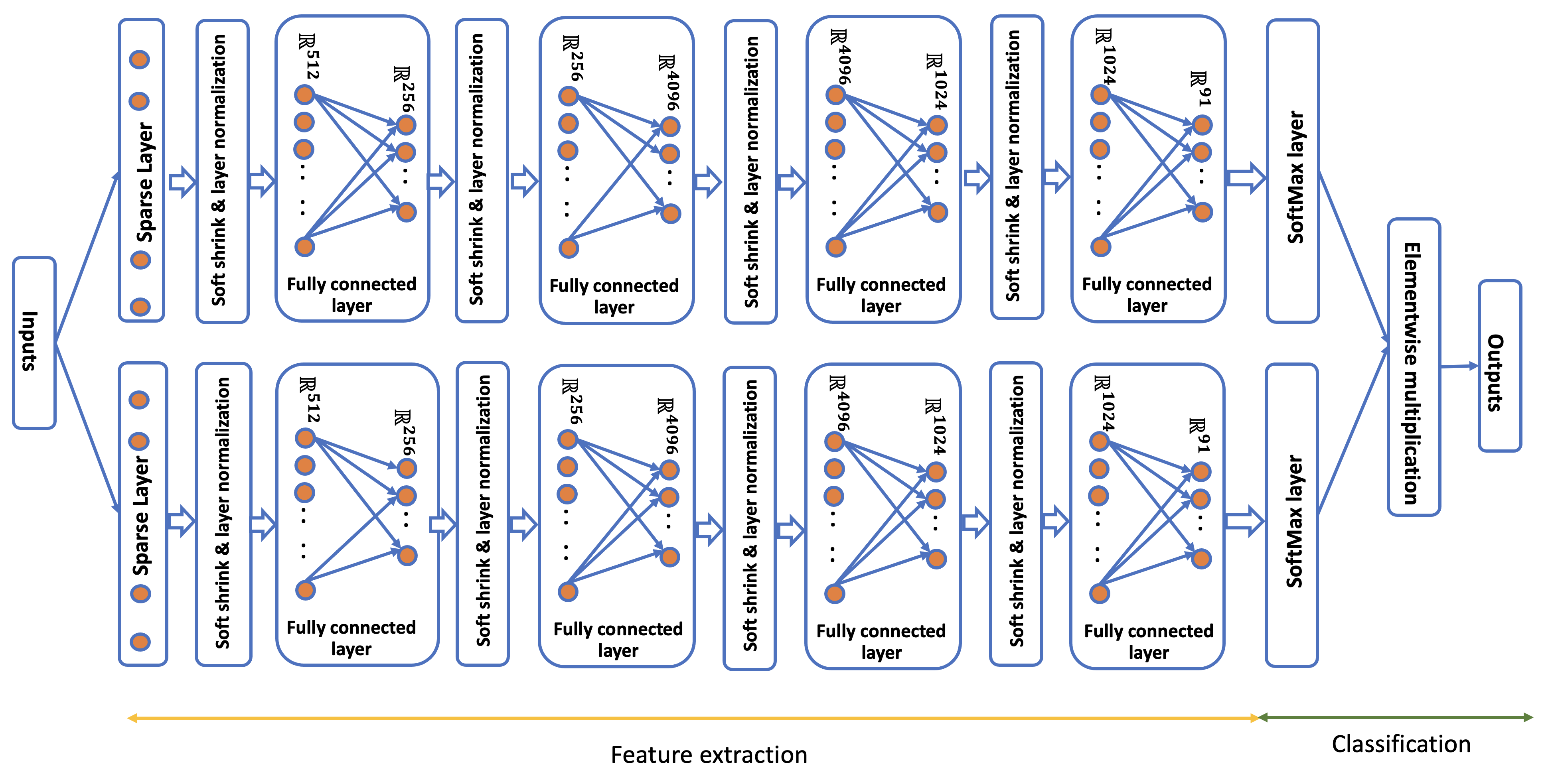
The multilayer sparse perceptron architecture and development (NNmod)
The design of the multilayer sparse perceptron is shown in Figure S1. This design is based on two primary assumptions, (i) the methylation data from central nervous system tumors and normal brain is embedded in low dimensional space, and (ii) random combinations of important probes can predict methylation class. The first assumption is typical of high dimensional data and is supported by examination of the singular value decomposition (SVD) of previously published reference data 6 (data not shown). In addition, the ability of combined methylation probes to predict methylation class is supported by previous implementations of random forest classifiers 6.
We constructed an 11-layer perceptron neural net. The input dimension is 51,108, composed of probes selected with feature extraction described in the network training section immediately following this section. The first layer is sparse, while the remaining ten layers are fully connected. The sparse layer maps 139,264 uniformly random sets of 256 features to the space [0,1]512. In other words, the sparse layer computes cosines of angles of vectors of length 256 drawn with uniform probability without replacement from the 51,108 input probes, which were selected by a LASSO model. This sparse feature layer can be considered a forest of random decision stumps 40, the output of which is fed through a standard perceptron.
Stochastic gradient descent was performed with a batch size of 32 on logarithms of output scores from the network using a learning rate of 0.001. These gradient descent parameters were obtained via a random search of the parameter space. The log-likelihood loss was minimized over the three-fold cross-validation. Using the evaluation partitions from the cross-validation splits, model calibration was performed with a multinomial logistic regressor. The final model was trained on the complete 2,801 samples using identical parameters following cross-validation.
Classifier cross-validation. To reduce the overfitting problem when training classifiers on high-dimensional data, all classifiers were cross-validated based on 1000 leave-out-25% cross-validations. We randomly selected 75% of the data used to train the classifiers (GSE90496), while the remaining 25% of the data were used for predictions. Stratified random sampling was performed for each methylation class or family to ensure the number of categories remained the same in each iteration. This validation process was repeated 1000 times (Fig. S2).
Model calibration. Calibration of machine learning methods may be necessary because the scores output by the classifier may have different scales when broken down by class, even when the scores are normalized so that they sum to 1. This poses problems for comparing the uncertainty in class or family calls between cases or even in the same case. Thus, the scores must be rescaled to form a well-calibrated multinomial distribution with minimal differences between expected values and variances between the class call groups.
Both RF and NN models were calibrated with the same multinomial logistic regression approach described by 6. The glmnet package (v-4.1-3) 41 was used with R bindings for the random forest and python bindings for the neural net.
Model robustness. To test whether missing methylation probes (features) affect our machine learning models, we randomly dropped 10% of the probes from the testing data (GSE109379 and SJCRH) and calculated the accuracy. The same probes at each round were used for all models. This process was repeated ten times to create 10 different missing sets of probes. Pearson’s correlation and Theil’s U uncertainty coefficients were computed using the ggpubr R package (v.0.4.0) and the DescTools R package (v.99.44), respectively. Pearson’s correlation coefficients with p-values were calculated to examine the linear relationship between the two sets of prediction scores (with and without missing probes). Theil’s U uncertainty coefficients were calculated to measure the nominal association between the two sets of labels predicted by the three classifiers on samples of GSE109379 and SJCRH data with and without missing probes.
Purity analysis. We performed an in silico simulated impurity experiment using different fractions of control and positive samples in GSE109379 and SJCRH test sets. The experiment was performed based on m-values. The in silico mixed m-values (mmixed) for each positive sample were computed as follows
$${m}_{\text{m}\text{i}\text{x}\text{e}\text{d}}=(1-p){m}_{\text{t}\text{e}\text{s}\text{t}}+p{m}_{\text{c}\text{o}\text{n}\text{t}\text{r}\text{o}\text{l}}$$
where \({m}_{\text{t}\text{e}\text{s}\text{t}}\) is the input m-value from the positive samples, and \({m}_{\text{c}\text{o}\text{n}\text{t}\text{r}\text{o}\text{l}}\) is the average m-values of up to 19 appropriate control (i.e. normal) tissue samples in the test sets, p is the proportion of normal control tissues contaminated in a tumor sample (ranging from 0 to 1 with 0.05 increment). The control samples were selected based on their control methylation class corresponding to the methylation class tumor as described in Table S10. The final measurement of the mixed sample was then converted back to beta values for classifier inputs.
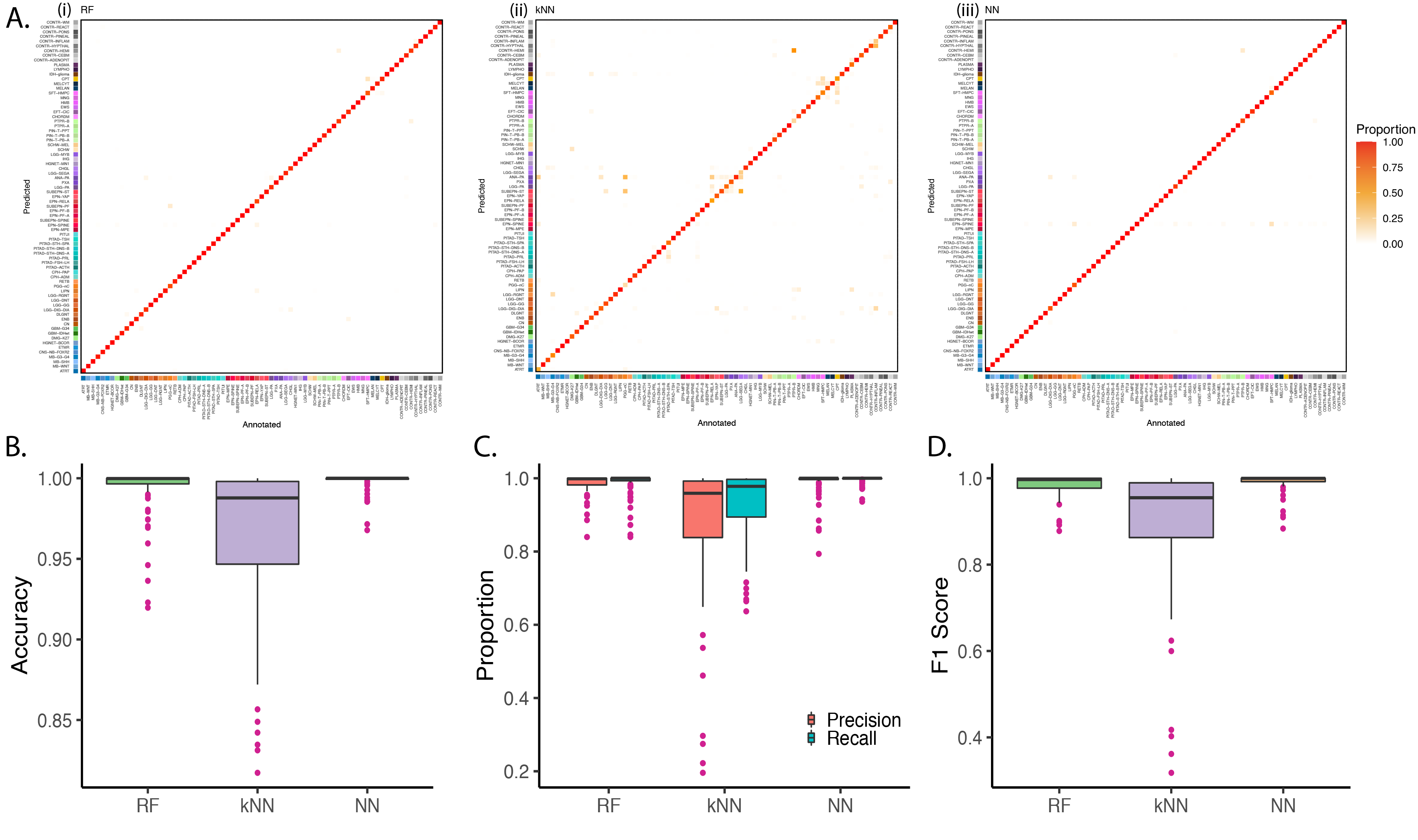
Model performance metrics. All models were evaluated based on accuracy, precision, specificity, recall, and F1 score. Classification accuracy is the number of correct predictions (true positives and true negatives) divided by the total number of predictions. Precision is the ratio of true positives to all the total positives predicted by a classifier. Specificity measures the proportion of true negatives correctly identified by a classification model. Recall or sensitivity is the ratio of true positives to all the ground truth positives. The F1-score is the harmonic mean of precision and recall and a good metric to measure the results in imbalanced classification problems. The higher the F1 score, the better the performance of a model. All measurements were computed using the caret R package (v.6.0–90).
{kind=link}
{kind=link}
{kind=link}