Bacterial strains and culture conditions
Acinetobacter baumannii Ts 50 − 16 and Klebsiella pneumoniae F 104 − 14 clinical isolates (collection of the N.F. Gamaleya Federal Research Center for Epidemiology and Microbiology, Ministry of Health of the Russian Federation), were implicated in the present study as biofilm-forming Gram-negative pathogens of the ESKAPE group. A. baumannii Ts 50 − 16 was isolated from patient’s sputum, intensive care unit, and K. pneumoniae F 104 − 14 was obtained from patient’s sputum, outpatient hospital. Both strains were stored at − 80°C and cultivated in pre-autoclaved Tryptic Soy Broth (TSB; SIFIN, Berlin, Germany) at 37°C, at 250 rpm overnight before performing further tests.
Recombinant expression and purification of proteins
Recombinant endolysins LysAm24, LysAp22, LysECD7, and LysSi3 were obtained as described previously (Vasina et al., 2021). In brief, proteins modified with polyhistidine-tag (8 His) were recombinantly expressed in Escherichia coli BL21(DE3) pLysS strain using 1mM β-D-1-thiogalactopyranoside induction at 37°C for 3 h. The cells were harvested by centrifugation (6,000×g for 10 min at 4°C) and incubated with 100 µg/mL lysozyme in lysis buffer (20 mM Tris-HCl, 250 mM NaCl, and 0.1 mM EDTA, pH = 8.0), and disrupted by sonication. Soluble proteins were purified on an NGC Discovery™ 10 FPLC system (Bio-Rad, Hercules, CA, U.S.) with a HisTrap FF column (GE Healthcare, Munich, Germany) pre-charged with Ni2+ ions. The filtered lysate was mixed with 30 mM imidazole and 1 mM MgCl2 and loaded on the column pre-equilibrated with binding buffer (20 mM Tris–HCl, 250 mM NaCl, and 30 mM imidazole, pH = 8.0). The fractions were eluted using a linear gradient to 100% elution buffer (20 mM Tris–HCl, 250 mM NaCl, and 500 mM imidazole, pH = 8.0). Resulting protein fractions were dialyzed against 20 mM Tris–HCl buffer (pH = 7.5).
The purity of endolysins was determined by 16% SDS-PAGE, and protein concentrations were measured using a spectrophotometer (Implen NanoPhotometer; Implen, München, Germany) at 280 nm and calculated using the predicted extinction coefficients [0.840, 0.831, 1.460, and 1.029 (mg/mL)−1 cm− 1 for LysAm24, LysAp22, LysECD7, and LysSi3, respectively]. The isoelectric points of endolysins LysAm24, LysAp22, LysECD7, and LysSi3 were predicted using the internet-source http://isoelectric.org.
Biofilm composition and morphology
Macrocolony assays were conducted on solid culture medium with application of three different staining techniques and without any dyes as an intact control. For the formation of BFs, cells from overnight cultures were harvested by centrifugation (6,000×g, 10 min, RT), resuspended in PBS (pH = 7.4), and diluted to achieve a cell density of approximately 3×106 CFU/mL. To obtain dual-species biofilms, the appropriate bacterial suspensions were mixed in a 1:1 (v/v) ratio. Thereafter, 7 µL of the suspension was inoculated onto solid TSB agar medium on 90 mm Petri dishes and BFs were grown at 37°C for 48 h without agitation. All images of colonies were acquired using HD automatic colony counter Scan 300 and provided software (Interscience, Saint-Nom-la-Bretèche, France).
Alcian blue 8GX 0.3% stain (PanReac AppliChem, Darmstadt, Germany) in a 4% acetic acid aqueous solution (pH = 2.5) was prepared to visualize polysaccharides with abundant carboxyl groups. Five mL of pre-filtered Alcian blue stain were carefully dripped on plates containing macrocolonies and then incubated for 15 min at RT without agitation. Subsequently, the plates were rinsed with 5 mL of deionised distilled water.
The results were confirmed by the Congo red stain absorption test (Freeman et al., 1989). For this, TSB agar was supplemented with pre-filtered 0.08% Congo red stain (Sigma, U.S.) to prepare agar plates. The 48-h old biofilms were cultured on the medium at 37°C without agitation. The intensity of red staining was considered proportional to the quantity of extracellular polysaccharides and proteins (Ahmad et al., 2020).
The morphotyping assay was conducted according to (Römling et al. 1998) with modifications. In brief, TSB agar was supplemented with a CRCB aqueous mixture, made up of pre-filtered 0.04% Congo red stain and 0.02% Coomassie blue G-250 stain (VWR (Avantor), Radnor, U.S.) to prepare agar-containing Petri dishes. Biofilms were grown on these CRCB agar plates for 24, 48, and 144 hours.
Microtiter biofilm formation assay (CV Mtp)
Overnight bacterial cultures in TSB were harvested (6,000×g, 10 min, RT), then suspended in PBS (pH = 7.4), diluted in the buffer to achieve a cell density of approximately 3×106 CFU/mL. Next, 100 µL of the suspension was added to sterile wells of a 96-well polystyrene cell culture plate, and incubated for 48 h at 37°C and 200 rpm. To obtain dual-species biofilms suspensions of both bacteria were mixed in a 1:1 (v/v) ratio. The wells’ content with planktonic cells was discarded, and the plate was washed three times with 200 µL of PBS (pH = 7.4), then air-dried for approximately 20 min. The dried BFs were stained with a 0.1% aqueous solution of Crystal violet (CV) for 15 min, at RT, followed by triple rinsing with water. The stained content was resolubilized in 200 µL of 33% acetic acid, and OD590 of the obtained solutions was measured using SPECTROstar NANO spectrophotometer (BMG LABTECH, Ortenberg, Germany). All experiments were performed with quadruplicate technical replicates and repeated at least in two independent assays. The values were normalized by dividing them by the OD590 of biofilm treated with the blank buffer. The level of biofilm formation was interpreted according to (Stepanović et al. 2007).
Quantification of eDNA in the biofilm
A measurement of eDNA was carried out with DNAse I (PanReac AppliChem, Darmstadt, Germany) treatment of biofilms, grown for 24 and 48 h. BFs were grown as described in the subsection “Microtiter biofilm formation assay” and treated with 100 µL of DNAse I solution (20 µg/mL of DNAse I in 20 mM Tris-HCl buffer, 1 mM CaCl2 (pH = 7.5)) or the same volume of the buffer at 37°C and 200 rpm, for 2 h. Thereafter, biofilms were rinsed with water, air-dried, and stained with 0.1% Crystal violet, following the OD590 measurement. The eDNA content was calculated as the difference in BF staining with and w/o of DNAse I treatment (BF biomass reduction).
Quantification of proteins content in the biofilm
A measurement of proteins’ quantity was conducted for 24- and 48-h old biofilms. Proteinase K (PanReac Applichem, Darmstadt, Germany) in 20 mM Tris-HCl buffer supplemented with 100 mM NaCl, 1 mM CaCl2 (pH = 7.5), at 50 and 800 µg/mL concentration was added to A. baumannii biofilms; and at 50, 800 and 1600 µg/mL concentration was added to K. pneumoniae biofilms. Preformed biofilms were treated with 100 µL of Proteinase K solutions or blank buffer at 37°C and 200 rpm, for 2 h. Thereafter, treated biofilms were rinsed, air-dried, stained with Crystal violet and analyzed as described in the subsection “Quantification of eDNA in the biofilm”.
Antibiofilm activity assessment
Mono-species and dual-species biofilms grown for 48 h and prepared for microtiter assay, were treated with either 100 µL of endolysin solutions at concentrations of 100 or 1000 µg/mL, or equal volume of 20 mM Tris-HCl (pH = 7.5) buffer as a negative control, for 2 h at 37°C and 200 rpm. After incubation, biofilms were rinsed twice, air-dried, stained with 0.1% Crystal violet, rinsed again, dissolved and analyzed as was described in the subsection “Microtiter biofilm formation assay”.
Microscopy of biofilms
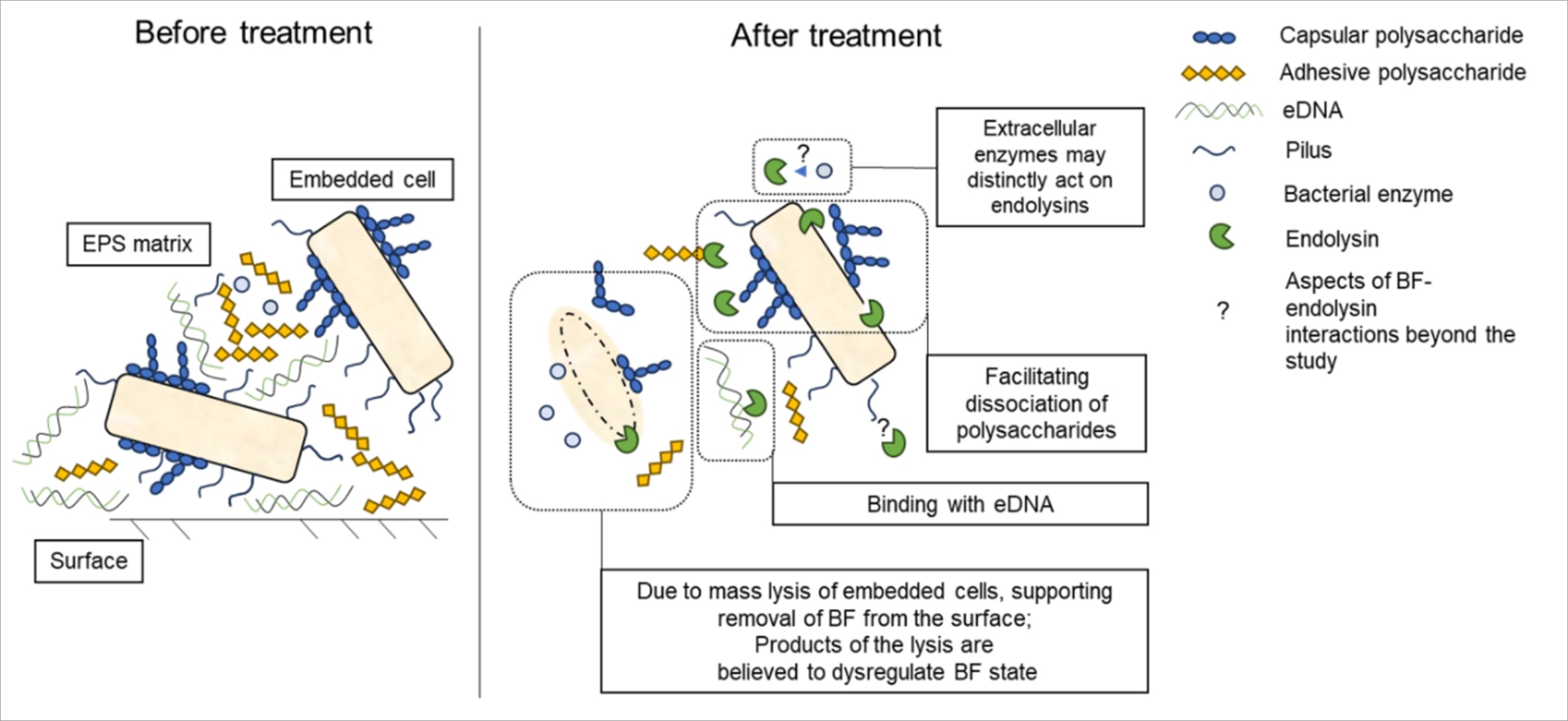
Sterile glass coverslips (Hampton Research, Aliso Viejo, CA, U.S.) were plunged into the overnight cultures, diluted in fresh TSB medium on Petri dishes and incubated at 37°C for 48 h without shaking. The slides were then carefully washed three times with sterile distilled water and air-dried. Two slides were treated with 300 µL of 20 mM Tris–HCl (pH = 7.5) control buffer, other pairs of slides were exposed to 300 µL of 100 µg/mL LysAm24, LysAp22, LysECD7, or LysSi3 solutions for 2 h at RT. Afterward, all slides were again washed with water two times. Air-dried slides were stained with a 0.1% aqueous solution of Crystal violet for 15 min at RT. All stained samples were rinsed once with water. Then, half of the slides were immediately rinsed twice and air-dried for microscopy. Another half of the slides were negatively stained to identify acid polysaccharides, which are crucial for antibacterial tolerance in most bacterial capsules and biofilms. A negative stain was performed using the Anthony method (Hughes and Smith 2013) with modifications. Following the method, CV-stained slides were submerged into 20% aqueous CuSO4 for 10 s, rinsed thoroughly with water, and then dried. All slides were imaged using Axiostar Plus Transmitted Light Microscope (Zeiss AG, Jena, Germany) at ×630 magnification.
DNA-binding effect of the endolysins
To detect interaction between eDNA and the endolysins we obtained genomic DNA of A. baumannii using a modified CTAB method (Minas et al. 2011). Briefly, cells from an overnight bacterial culture were harvested, once washed, and solubilized in a CTAB solution (2% w/v cetrimonium bromide (Helicon, Moscow, Russia), 100 mM Tris-HCl, 20 mM EDTA and 1.4 M NaCl, pH = 8.0), ex tempore supplemented with 0.2% β-mercaptoethanol, at 65°C for 40 min. Then, the suspensions were mixed with an equal volume of chloroform-isoamyl alcohol (24:1), and the aqueous phase was collected after centrifugation at 12,000×g for 10 min, RT. The DNA precipitation proceeded in 0.6 volume of isopropanol overnight at – 25°C. The precipitates were harvested at 8,000×g for 15 min and washed three times with 80% ethanol. After ethanol was discarded, the pellets were suspended in 20 mM Tris-HCl (pH = 7.5) buffer with 1 µg/mL RNAse A (PanReac Applichem, Darmstadt, Germany), incubated for 15 min at 37°C, afterwards RNAse A was inactivated at 65°C for 15 min. Quality of the DNA samples was estimated spectrophotometrically, analyzing A260/230 and A260/280, thereafter a concentration of DNA was measured using Qubit DNA HS Assay Kit and Qubit 3.0 fluorometer (Thermo Fisher Scientific Eugene, Oregon, U.S.).
Endolysins LysAm24, LysAp22, LysECD7, and LysSi3 solutions in 20 mM Tris-HCl buffer at 10 or 50 ng/µL concentration were mixed with DNA solutions (at a concentration of 20 ng/µL) at a protein:DNA (P:DNA) mass ratio of 2:1 or 10:1 (final volume of the mixtures was 7.5 µL) and incubated for 30 min at RT. Solutions containing either DNA or investigated endolysin in 20 mM Tris-HCl buffer (pH = 7.5) were used as control. The influence of electrostatic interactions was assessed by adding NaCl to final concentrations of 150 or 300 mM to the mixtures (P:DNA w/w ratio of 10:1) during their incubation. Post-incubated solutions were mixed with the 6X DNA Loading Dye (Thermo Fisher Scientific Eugene, Oregon, U.S.) and loaded in 1% agarose gel, containing 0.2 µg/mL ethidium bromide (Helicon, Moscow, Russia). An electrophoretic separation of all samples was performed in the Tris-borate buffer (50 mM Tris-base, 50 mM boric acid, 2 mM EDTA, pH = 8.3) at 80 V for an hour. Imaging was performed using the Gel Doc EZ gel documentation system (Bio-Rad, Hercules, CA, U.S.).
To correlate the binding assay with the inhibitory effect of eDNA, an antibacterial activity test was performed. An overnight bacterial culture of A. baumannii was diluted 30-fold in LB broth and grown to the exponential phase (OD600 = 0.6). Subsequently, the cells were harvested by centrifugation (6,000×g, 10 min) and resuspended in the same volume of PBS (pH = 7.4). Each suspension was diluted in the 20 mM Tris-HCl (pH = 7.5) to a final density of approximately 106 cells/mL. Afterward, 100 µL of the bacterial suspensions and 100 µL of the pre-incubated protein-DNA solutions (a final protein concentration was 10 µg/mL, a final DNA concentration was 1 or 5 µg/mL) were mixed in 96-well plates. Buffers with or without DNA were used as the negative controls. The mixtures were incubated at 37°C for 30 min with shaking at 200 rpm and then were diluted 10-fold in PBS (pH = 7.4). Then, 100 µl of each dilution were plated onto LB agar, and the number of bacterial colonies were counted after an overnight incubation at 37°C. All experiments were performed in quadruplicate, and the antibacterial activity was expressed as follows: Antibacterial activity (%) = 100% – (CFUexp/CFUcont) × 100%, where CFUexp is the number of bacterial colonies in the experimental culture plates, and CFUcont is the mean number of bacterial colonies in the control culture plates.
Statistical Analysis
The data were analyzed and illustrated using GraphPad Prism 9.0 software. According to the results of normality tests (Kolmogorov-Smirnov’s method), data sets were compared using appropriate statistical tests with corrections for multiple comparisons (detailed information on the chosen analysis methods is provided in captions and results).
{kind=link}