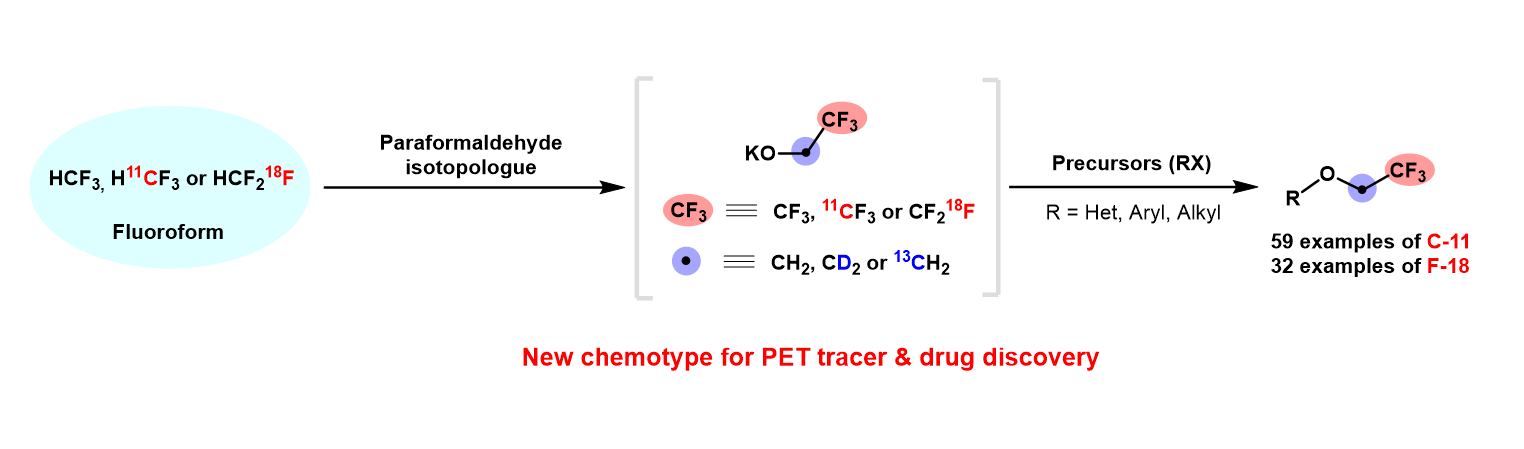
Nucleophilic addition of fluoroform to aldehydes to produce carbinols is well-known (40). However, the reaction of fluoroform with the simplest aldehyde, formaldehyde, has not been explored. We considered that this reaction should produce a very useful reagent, 2,2,2-trifluoroethoxide. Because of the toxicity and volatility of formaldehyde (41), we decided to explore solid paraformaldehyde as an alternative for producing 2,2,2-trifluoroethoxide. Treatment of fluoroform as limiting reagent with a mixture of paraformaldehyde and t-BuOK in DMF produced 2,2,2-trifluoroethanol in 41% yield after water quench (Table S1, entry 1). Under optimal conditions, fluoroform was converted into 2,2,2-trifluoroethanol in 88% yield within 30 minutes (Table S1, entry 13). Increasing the reaction time did not improve the conversion appreciably (Table S1, entries 11 and 12). Other bases or solvents adversely affected yield (Table S1, entries 14–18).
We next tested the reactivity of the putative intermediate, potassium 2,2,2-trifluoroethoxide, by treatment with 2-chloropyrimidine. To our delight, reaction proceeded smoothly at room temperature to afford 2-(2,2,2-trifluoroethoxy)pyrimidine (1) in 71% yield within 2 hours (Fig. 2A). However, treatment of potassium 2,2,2-trifluoroethoxide with 1-fluoronaphthalene gave only trace 1-(2,2,2-trifluoroethoxy)naphthalene (2), even at elevated temperature. We considered that an aryl hypervalent iodine center in an aryliodonium ylide might show more reactivity. Indeed, treatment of potassium 2,2,2-trifluoroethoxide with naphthalen-1-yl iodonium-(2,2-dimethyl-1,3-dioxane-4,6-dione)ylide at 60 oC produced 2 in 63% yield (Fig. 2A). Thus, gratifyingly, we had succeeded in converting stoichiometric amounts of fluoroform into a useful synthon, potassium 2,2,2-trifluoroethoxide in high yield and in showing the utility of this synthon for trifluoroethoxylation of homoarene and heteroarene under mild conditions. We anticipate that this new metal-free transformation can find wide application in fluorine chemistry. Indeed, we readily produced several trifluoroethoxy compounds in high yields by this chemistry as standards for use in the remainder of this study (see Supporting Information).
We next aimed to explore this synthetic methodology for robust broad-scope radio-trifluoroethoxylations as a potentially new route to PET tracers. For this purpose, we routinely [11C]fluoroform by CoF3-mediated fluorination of cyclotron-produced [11C]methane (22). Treatment of [11C]fluoroform (37–296 MBq) with a mixture of t-BuOK (50 µmol) and paraformaldehyde (17 µmol) in DMF for only 3 minutes at room temperature gave an excellent yield (88%) of 11CF3CH2OH upon hydrolysis of the putative intermediate (Fig. 2B, entry 1). Increasing the reaction time to 5 minutes only slightly increased the yield. A larger quantity of paraformaldehyde (50 µmol) afforded 11CF3CH2OH in 97% yield. The yield of 11CF3CH2OH was quantitative when [11C]fluoroform was treated with 1: 2 molar mixture of paraformaldehyde and t-BuOK for 4 minutes (Fig. 2B, entry 5). These reaction conditions were therefore deemed optimal. This success encouraged us to test the efficacy of 11CF3CH2OK for introducing the 11C-trifluoromethyl moiety into a wide range of compounds.
First, we examined the reactivity of 11CF3CH2OK towards heteroarenes. To our delight, 11CF3CH2OK reacted at room temperature to produce a wide range of desired 11C-2,2,2-trifluoroethoxy heteroarenes in moderate to excellent yields within just 1 minute (Fig. 3A). Attractive features to emerge from this labeling protocol were: 1) the 11CF3CH2OK, can be used without isolation; 2) reaction conditions are mild and rapid; 3) substrate scope is broad, and encompasses pyridines, pyrimidines, pyrazine, thiazoles, triazines, quinolines, and isoquinolines; 4) in addition to halides (F, Cl, and Br), leaving groups such as nitro ([11C]4) and methyl sulfone ([11C]9) are highly effective; 5) functional group tolerance is high, with aldehyde ([11C]3), bromo ([11C]4, [11C]6), nitrile ([11C]5), methoxy ([11C]7), and Boc protection ([11C]9) all well tolerated. Heteroarenes having 1 to 3 nitrogen atoms ([11C]1, [11C]3–[11C]14) were compatible with the reaction conditions and afforded the desired 11C-labeled products in acceptable yields (21–94%). Furthermore, the late-stage 11C-trifluoroethoxylation of complex biomolecules was highly effective as shown by the labeling of analogues of several drug-like compounds [Imiquimod ([11C]15), Milrinone ([11C]18)], drug precursors [Erlotinib ([11C]19), Canagliflozin ([11C]20), Pazopanib ([11C]21), Palbociclib ([11C]22)], the herbicide Clopyralid ([11C]17), and a purine derivative ([11C]16)] in moderate to very good yields. Notably, 11C-trifluoroethoxylation occurred preferentially at the more electron-deficient site (e.g., the ortho vs meta pyridinyl site for [11C]17) or aryl ring (e.g., the pyridinyl vs homoarene ring in [11C]6 and [11C]20).
We found that homoarene precursors with common leaving groups were more challenging for 11C-trifluoroethoxylation than the reactive heteroarenes (42). We anticipated that the use of a more powerful hypervalent aryliodonium leaving group (43, 44) could alleviate this issue. We opted to explore this possibility for the one-pot 11C-trifluoroethoxylation of homoarene substrates. First, we screened conditions for the reaction of [11C]potassium 2,2,2-trifluoroethoxide with iodonium ylide derived from Meldrum’s acid (naphthalen-1-yl(2,4,6-trimethoxyphenyl)iodonium tosylate) (Table S3). We found that treating a mixture of [11C]CF3CH2OK with the iodonium salt precursor (55 µmol) in DMF at 60 oC for 3 minutes gave a high and optimal yield of the desired [11C]2 (87%; Table S3, entry 2). The 2,4,6-trimethoxyphenyl group served as an effective aryl spectator ring; no [11C]1,3,5-trimethoxy-(2-(2,2,2-trifluoroethoxy))benzene, was produced. An increase in temperature did not improve the yield of [11C]2. Reduction in precursor amount reduced yield. Given the high yield obtained for [11C]2 with this approach under optimal conditions, we proceeded to explore substrate scope. Substituent electronics had substantial influence on reaction yields ([11C]23–[11C]32). Electron-withdrawing groups in ortho and para position gave high yields for the 11C-trifluoroethoxylation (Fig. 3B). 11C-Trifluorethoxylation yields were lower for substrates with para- electron-donating or meta- electron-withdrawing groups. Novel cross-coupling synthons, [11C]24–[11C]27, were obtained in useful yields. We drew attention to the syntheses of [11C]26 and [11C]28, where mesityl was used as the partner aryl ring in the iodonium salt precursor. 11C-Trifluoroethoxylation was directed to the other aryl ring. This is an interesting observation because here ring chemoselectivity is opposite to that seen for the non-copper mediated radiofluorination of aryl(mesityl)iodonium salts (45).
We also explored aryliodonium ylides as precursors. 11C-Trifluoroethoxylation of three model ylides gave [11C]33–[11C]35, in moderate to high yields. Moreover, [11C]36, an analogue of the antidiabetic drug emplagliflozin (®Jardiance) was also obtained in moderate yield (47%) from an ylide. These are the first examples of iodonium ylides serving as precursors for the trifluoroethoxylation reaction. In addition, two activated fluoroarenes, with ortho electron-deficient aryl rings, gave [11C]37 and [11C]38 in moderate to good yields where fluoride was the leaving group.
Fluorine-18 labeling of PET tracers at aliphatic carbon by nucleophilic substitution of a good leaving group with [18F]fluoride (19) can often lead to an [18F]fluoroalkyl group that is vulnerable to radiodefluorination in vivo and to accumulation of [18F]fluoride ion in the bone including skull. This can hamper accurate quantification of tracer uptake, especially in brain (20, 46, 47). 18F-Labeling in a 2,2,2-trifluoroethoxy group instead of an 18F-fluoroalkyl group could be a strategy to circumvent this issue. This consideration prompted us to investigate the reactivity of 11CF3CH2OK with aliphatic substrates. We started with a model compound, a precursor to Posaconazole (®Noxafil) with a tosylate leaving group to optimize precursor amount and reaction temperature (Table S4). 11C-Trifluoroethoxylation produced excellent yields of [11C]45 (89%) under conditions found to be optimal for hypervalent iodonium precursors (Table S4, entry 3). Again, yield did not increase with temperature (Table S4, entry 4). Reduction in precursor amount drastically diminished yield (Table S4, entries 5 and 6). We next tested reaction scope by attempting to prepare a range of 11C-labeled alkyl-2,2,2-trifluoroethyl ethers from aliphatic precursors, including fourteen 11C-labeled biomolecules (Fig. 4). [11C]41 was obtained from a long chain iodoalkyl precursor in acceptable yield (45%). Benzyl halide and α-chloroacetyl precursors were readily converted into their analogous 11C-2,2,2-trifluoroethoxy ethers ([11C]42, [11C]47, [11C]48, [11C]51, [11C]52) in high yields (53–89%) with good tolerance of other functional groups. Aliphatic tosylates derived from a variety of commercially available dug-like molecules (MCPA, Helional, Ketoconazole, Bendazac, an α-D-glucopyranoside derivative, Oxaprozin, Ospemifene, Pterostilbene, and Cyhalofop-butyl) reacted readily with 11CF3CH2OK to provide the desired 11C-2,2,2-trifluoroethoxy ethers, [11C]39, [11C]40, [11C]43–[11C]46, [11C]49, [11C]50, and [11C]53–[11C]56, in moderate to excellent yields (34–93%). Precursors with leaving groups attached to an ethylene glycol linker gave excellent yields of 11C-2,2,2-trifluoroethoxy ethers. Notably, aliphatic 11C-trifluoroethoxylation occurred in preference to reaction at aromatic sites.
We measured the molar activity for a model product [11C]1, produced by the 11C-trifluoroethoxylation of 2-chloropyrimidine, to verify that this labeling technique is no-carrier-added (NCA) and gives high molar activity. Starting with about 10 GBq of cyclotron-produced [11C]methane that has been produced from a 10 µA × 10 minute cyclotron irradiation, [11C]1 was obtained with a molar activity of 60 GBq/µmol, corrected to the end of radionuclide production (ERP). Such a high molar activity from a relatively limited cyclotron irradiation shows that the labeling reaction is invulnerable to carrier addition and dilution of molar activity (15, 39).
Having established an efficient route for 11C-trifluoroethoxylation, we next focused on translation of this new labeling method from carbon-11 to fluorine-18 with a few representative substrates. For this purpose, [18F]fluoroform was produced from no-carrier-added [18F]fluoride and difluoroiodomethane (48). CF218FCH2OK was generated by treatment of the [18F]fluoroform with paraformaldehyde and t-BuOK in DMF in greater than 95% yield. The reactivity of CF218FCH2OK was assessed under the optimal conditions found for 11C-trifluoroethoxylations (Fig. 5). 18F-Trifluoroethoxylations of aryl and aliphatic precursors proceeded smoothly and provided corresponding products in moderate to excellent yields similar to those from 11C-trifluoroethoxylation. Heteroarenes, such as pyridine, quinoline, pyrimidine, isothiazole, and 1,3,5-triazine, with halogen leaving groups were converted into the corresponding 18F-2,2,2-trifluoroethoxy ethers in high yields (82–96%; Fig. 5A). Dependency of labeling position on aryl ring position of the leaving group (e.g., ortho- vs meta- as in [18F]4 and [18F]17) or on the nature of the aryl ring (e.g., [18F]20) was as seen for 11C-labeling. Heteroaryl rings with more structural complexity and diverse functionality were conveniently labeled at room temperature within 5 minutes and produced the corresponding 18F-labeled compounds in excellent yields ([18F]15, [18F]17–[18F]22; 56–77%; Fig. 5A). These results indicate high potential for application of this labeling method to prospective structurally complex PET tracers. Homoarene precursors, including diaryliodonium salts ([18F]27 and [18F]31), aryliodonium ylides ([18F]35 and [18F]36), and fluoro precursors ([18F]37 and [18F]38), afforded useful yields of 18F-labeled products (27–69%; Fig. 5B), as for the11C-trifluoroethoxylations. Remarkably, the unprotected hydroxyl group in the Ataluren precursor was well tolerated ([18F]38) showing compatibility of this labeling protocol to sensitive functionality.
We were keen furthermore to know whether potassium 18F-2,2,2-trifluoroethoxide could be useful for labeling at aliphatic carbon, given the limited availability of methods for constructing stable alkyl-CF218F bonds (49). In this regard, we tested identical reaction conditions to those used for 11C-trifluoroethoxylation on diverse aliphatic substrates, prepared from drugs, herbicides, and other biomolecules. To our delight, this protocol successfully enabled the installation of a 18F-2,2,2-trifluoroethoxy moiety onto aliphatic carbon in a variety of complex structures in acceptable to excellent yields ([18F]44–[18F]46, [18F]49–[18F]56; 15–95%; Fig. 5C). Taken together, these results show that this new methodology, based on the transformation of fluoroform into potassium 2,2,2-trifluoroethoxide and subsequent functionalization of aliphatic carbons, is equally versatile for both carbon-11 and fluorine-18 with the same non-radioactive precursor.
Isotopologues differ only in their isotopic substitutions and play an important role in drug development (50). Deuteration (51) is widely practiced to improve the metabolic stability of PET tracers in 18F-fluoroalkyl positions. 13C-Labeling enables investigations of drug pharmacokinetics and metabolism by 13C-NMR spectroscopy and mass spectrometry (52, 53). Simple methods for accessing stable isotopically labeled compounds are highly desirable. The availability of isotopically labeled fluoroforms (H12CF3, H11CF3, and HCF218F) and paraformaldehyde [(12CH2O)n, (CD2O)n and (13CH2O)n] and our method for the in situ generation of CF3CH2OK from fluoroform, provide an opportunity to explore the incorporation of isotopically labeled 2,2,2-trifluoroethoxy groups into a diverse array of substrates. For demonstration, we synthesized isotopologues of 57, an analog of a well-known COX-1 PET tracer, [11C]PS13 (32). Compounds [11C]57 and [18F]57 were readily obtained by treating a tosylate precursor with [11C/18F]CF3CH2OK under optimized conditions. The reaction was equally effective when substituting paraformaldehyde with (CD2O)n and (13CH2O)n, leading to high yield syntheses of [2H]57, [2H/11C]57, [2H/18F]57, [13C]57, [13C/11C]57, and [13C/18F]57 (Fig. 6). Hence, this novel isotope labeling protocol has exceptional potential for broad application.
{kind=link}