3.1 Transcriptome analysis induced by metabolic stimulation
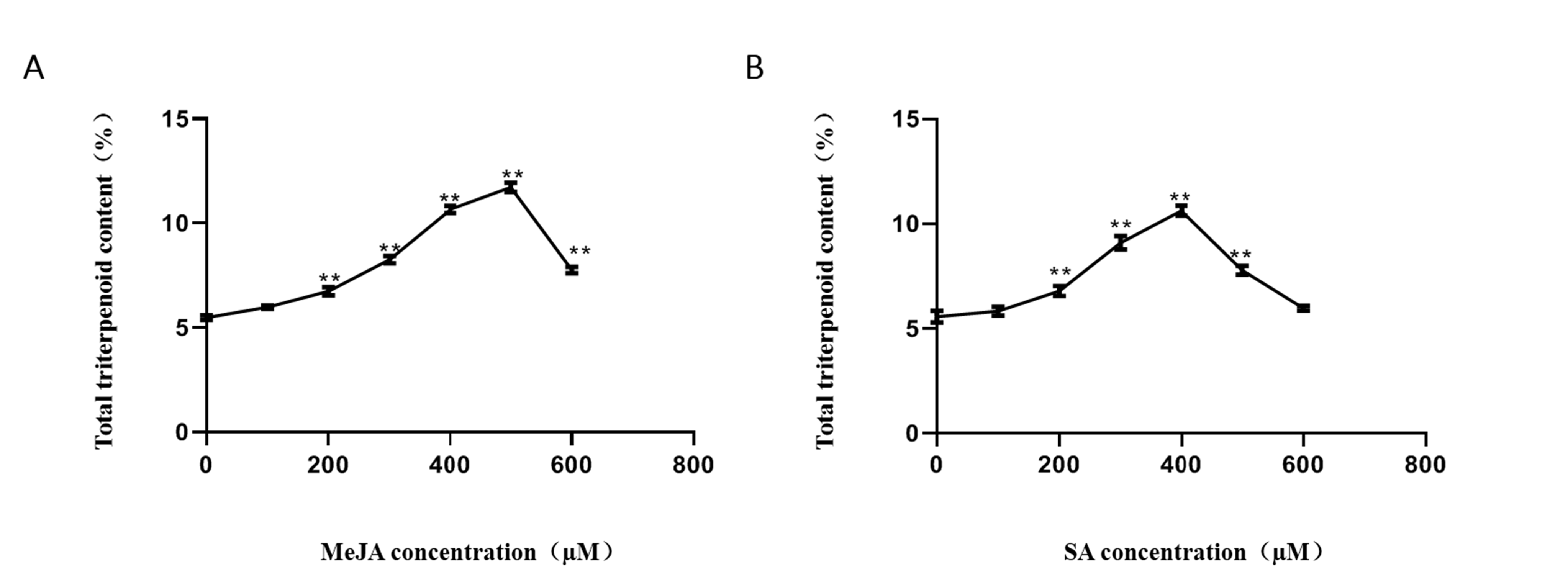
The optimal concentration of 500 μM methyl jasmonate (MeJA) and 400 μM salicylic acid (SA) were determined by single factor experiment (Figure S2). Consequently, the total triterpenoid content of samples from five groups was measured. The results showed elevated contents for total triterpenoids in the MD-, MeJA-, and SA-induced groups relative to the NC group (Figure S3), which was consistent with other research showing that synthesis of triterpenoids was affected by metabolic stimulation [36]. Then the RNAs of different groups treated by metabolic stimulation were sequenced with an Illumina high-throughput sequencing platform to obtain an overview of the transcriptome profile of H. angustifolia among the five groups. In all, 121.62 Gb high-quality data (at least 6.91 Gb for each sample and 23.46 Gb for each group) with 43.67%, GC contents were generated from 15 samples. The raw data and quality control data are characterized in Table S4. Then 581,934 transcripts and 424,824 unigenes were obtained after trimming and assembly of the clean data. The mean size of the unigenes was 655 bp, with a N50 length of 1,300 bp (Table 2).
Table 2 Analysis of assembly transcriptome
|
Item
|
Number
|
|
Total sequence base
|
381,166,620
|
|
Total transcripts num.
|
581,934
|
|
Total unigenes num.
|
424,824
|
|
Largest (bp)
|
16,851
|
|
Smallest (bp)
|
201
|
|
Length N50 (bp)
|
1,294
|
|
Mean Length(bp)
|
655
|
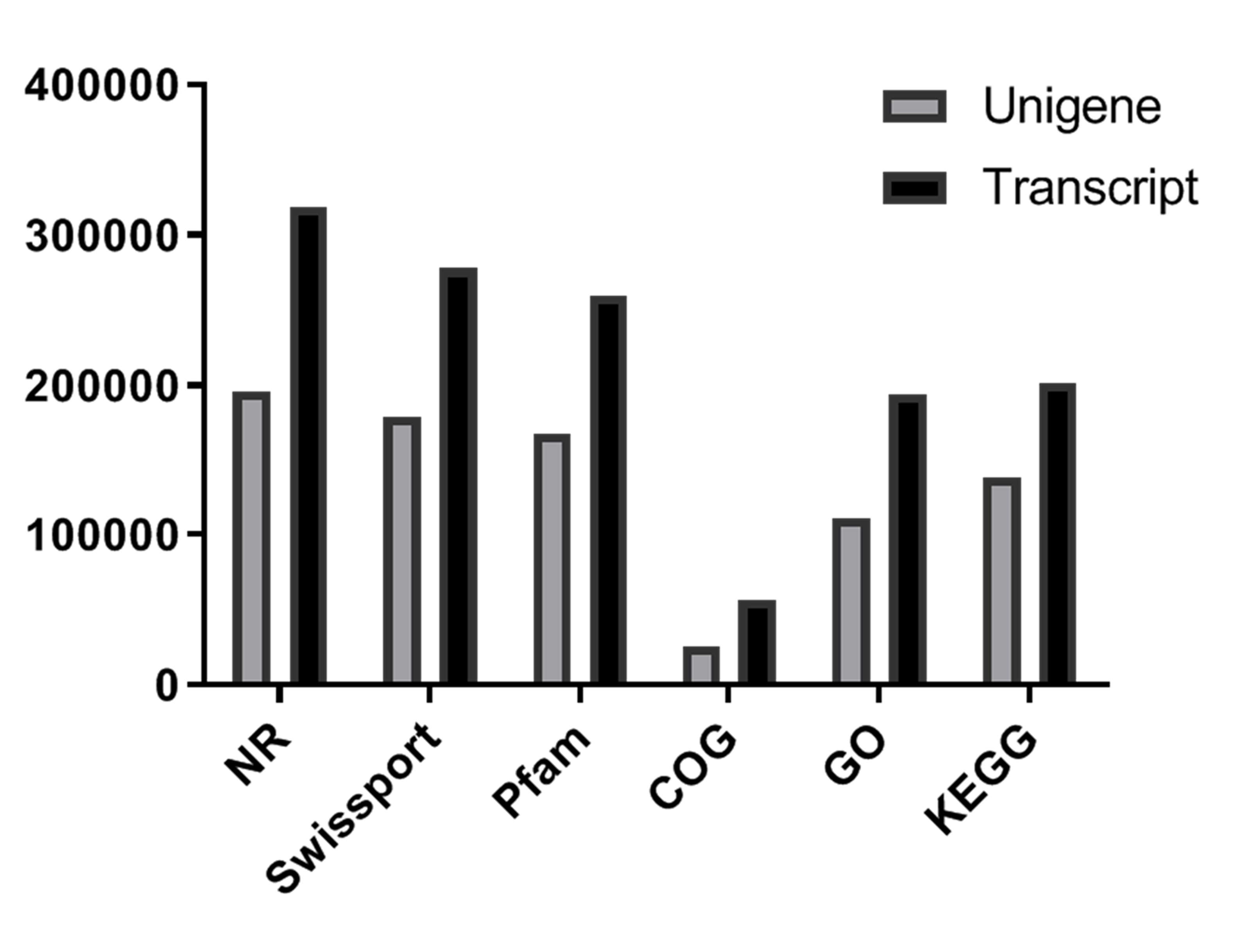
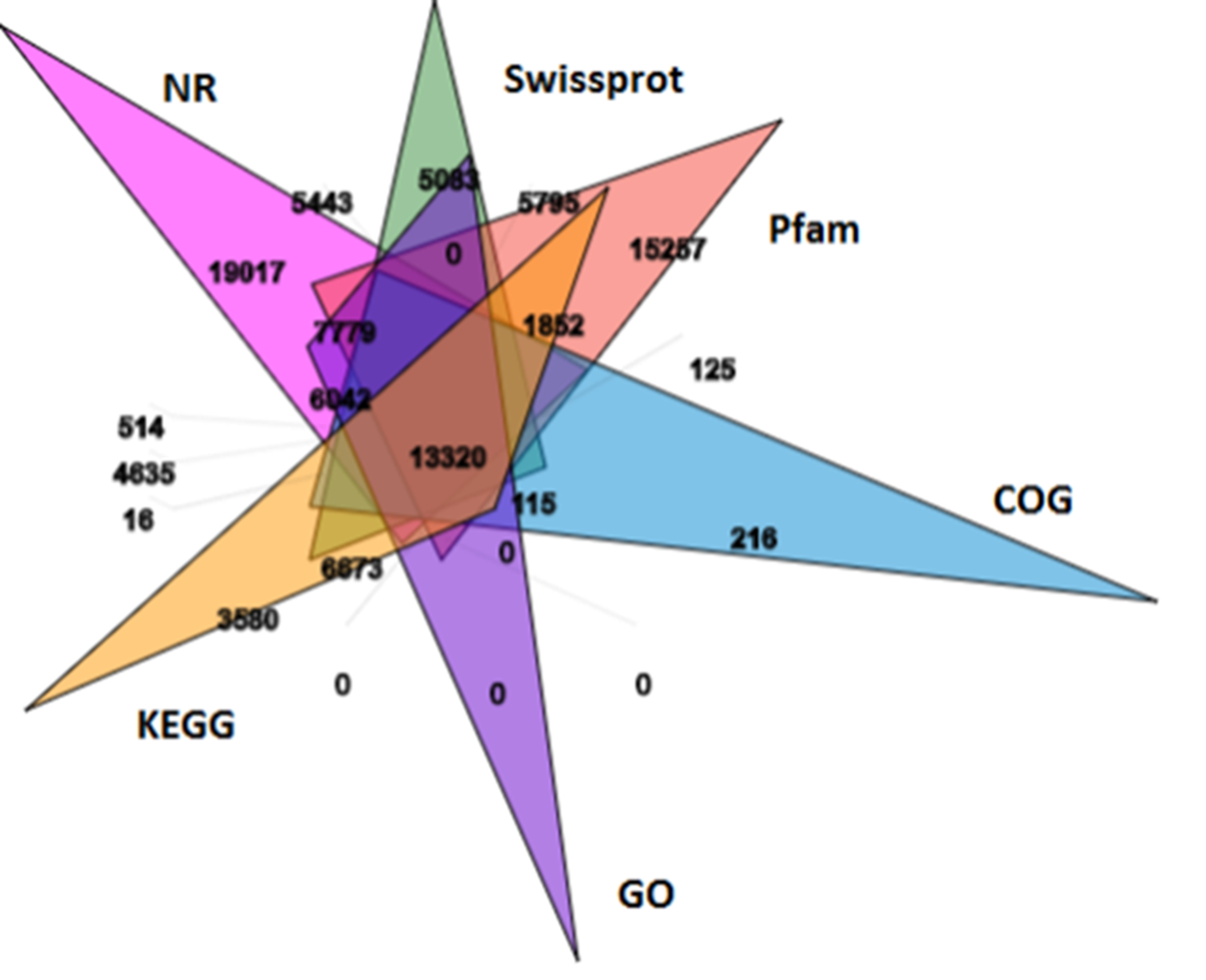
Sequence annotation was performed using the BLAST algorithm against six public databases. In all, 196,252, 179,036, 167,974, 25,390, 110,706, and 138,066 unigenes, respectively, were annotated in these databases (Figure S4, Table S5). In total, among the 424,824 unigenes generated by transcriptional sequencing, 245,709 (57.84%) received at least one hit in those databases, with 13,320 unigenes sharing common annotation (Figure S5).
3.2 Analysis of DEGs under different treatments
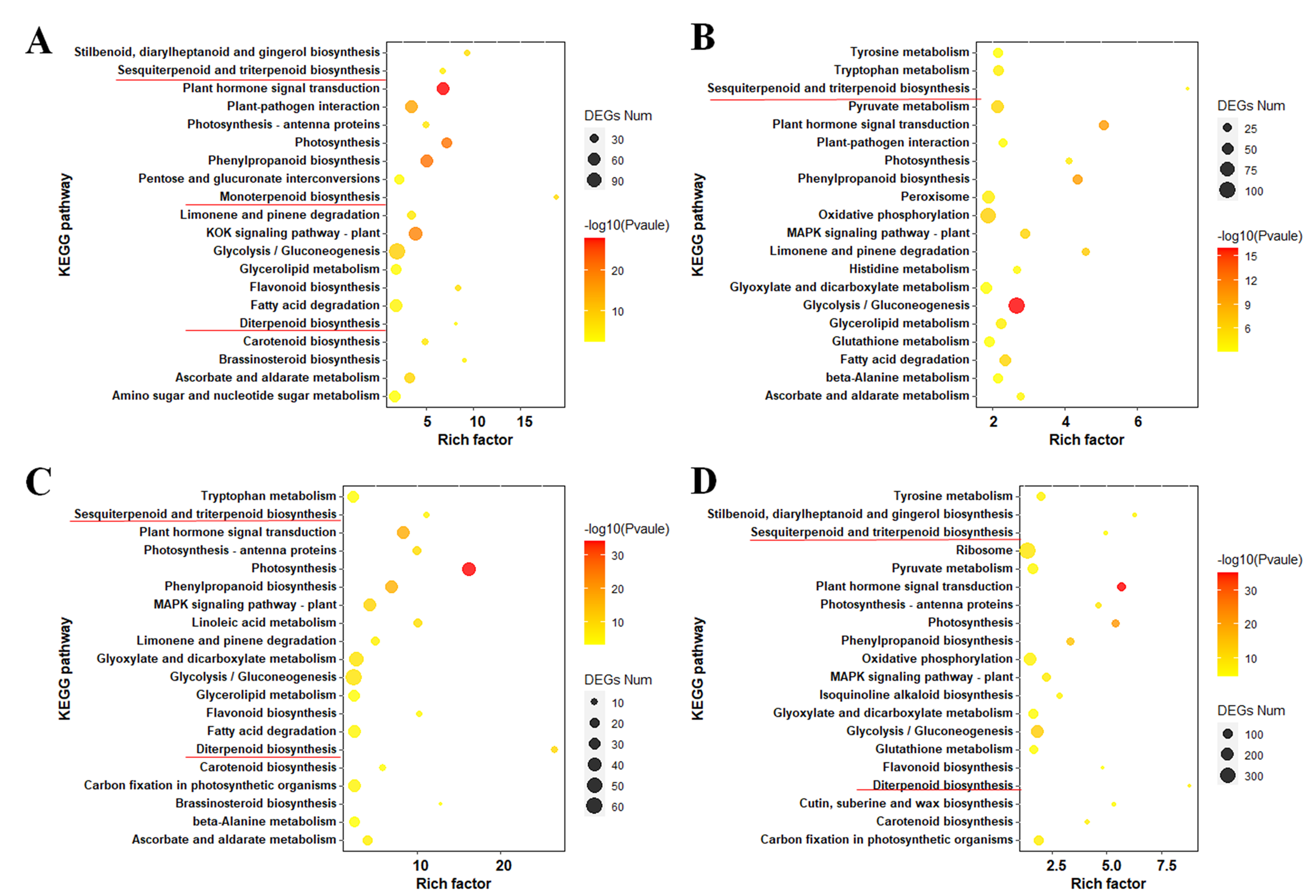
The transcriptome statistics of those five groups were compared pairwise to establish potential DEGs among the different groups. Compared to the NC group, there was the greatest amount of DEGs in the MD group (Figure 2A), and the number of up-regulated DEGs in each metabolic stimulation treatment group and EtOH group were higher than that of the downregulated genes (Figure 2B). The Venn diagram presented in Figure 2C indicates that 774 DEGs were shared among the four aforementioned pairwise comparisons, and a total of 22,430 DEGs were obtained among the four comparison groups. Heat maps were produced based on the expression patterns of all DEGs, and these could be divided into eight clusters (Figure 2D). KEGG enrichment analyses of DEGs in four pairwise groups(Figure S6, Table S6-9)suggested that pathways with the most enrichment were mostly associated with triterpenoid synthesis, revealing the expression of genes related to triterpenoid synthesis was upregulated after metabolic stimulation, leading to the enhancement of triterpenoid accumulation for resistance, which is consistent with the results of previous research [30, 37].
In addition, WGCNA was conducted to further investigate the potential genes involved in the triterpenoids biosynthesis of H. angustifolia. All DEGs were submitted to construct a scale-free co-expression network. The dynamic hierarchical tree algorithm was employed to divide cluster trees constructed by DEGs, resulting in 10 co-expression modules. These modules are named according to their colors, namely blue (1619 genes), brown (891 genes), turquoise (2505 genes), grey (17 genes), yellow (759 genes), pink (55 genes), green (673 genes), black (87 genes), magenta (52 genes), and red (115 genes) (Figure 3). The genes within each module were selected for enrichment in the KEGG pathway. Statistically significantly enriched genes (P < 0.05) were filtrated for deep analyses. Results showed that five modules (Blue, turquoise, black, green and magenta), consisting of 4,936 genes in total, were related to triterpenoid biosynthesis.
3.3 Quantitative real-time PCR (qRT-PCR) to verify the DEGs.
The expression patterns of 41 key candidate genes (Figure S7, Table S10) involved in triterpenoids biosynthesis were analyzed. Among them, 18 genes presented significantly different expression levels among five treatment groups, nine of them were chosen to analyze the expression level using qRT-PCR to further verify the reliability of our RNA-Seq data. As shown, the expression patterns of these nine DEGs were consistent with the RNA-Seq data (Figure 4), which confirmed the accuracy of our transcriptome data.
3.4 Cloning and sequence analysis of the candidate genes
To shed light on acylated triterpenoids biosynthesis in H. angustifolia, putative key enzymes encoding genes including three OSCs (named HaOSC1, HaOSC2 and HaOSC3), four CYP450s (named HaCYPi1, HaCYPi2, HaCYPi3 and HaCYPi4), two TATs (named HaTAT1 and HaTAT2), and one TBT (named HaTBT) were targeted for the functional analysis. Their nucleotide sequences were summarized and exhibited in Table S11 and physicochemical properties of corresponded proteins were documented in the Table S12, two-dimensional structure information of proteins was classified in the Table S13 and three-dimensional structures were displayed in Figure S8.
Phylogenetic analyses of three OSCs with a set of well characterized OSCs revealed that HaOSC1 and HaOSC3 were grouped into the lupeol synthase clade, and HaOSC2 was classified as a β-amyrin synthase (Figure 5A, Table S14). In addition, we found that all three candidate OSCs contained complete SQHop-cyclase-N and SQHop-cyclase-C domains. Furthermore, several conserve motifs were present in all three candidate OSCs, including DCTAE, MWCYCR, and QW repeat (Figure 5B).
The results of phylogenetic analyses also implied that four CYP450 candidate genes were well documented in the CYP716A subfamily (Figure 5C, Table S15), which primarily includes CYP450 enzymes with a C-28 oxidation function [38, 39](Fukushima et al. 2011; Suzuki et al. 2018). Analysis of conserved domains demonstrated that four candidate genes all had a complete Cytochrome P450 domain. Motif elucidation revealed that HaCYPi1, HaCYPi3, and HaCYPi4 possessed conserved motifs, including EXXXR, (A/G)GX(D/E)T and FXXGXRXCXGX, while HaCYPi2 lacks the motif FXXGXRXCXGX (Figure 5D).
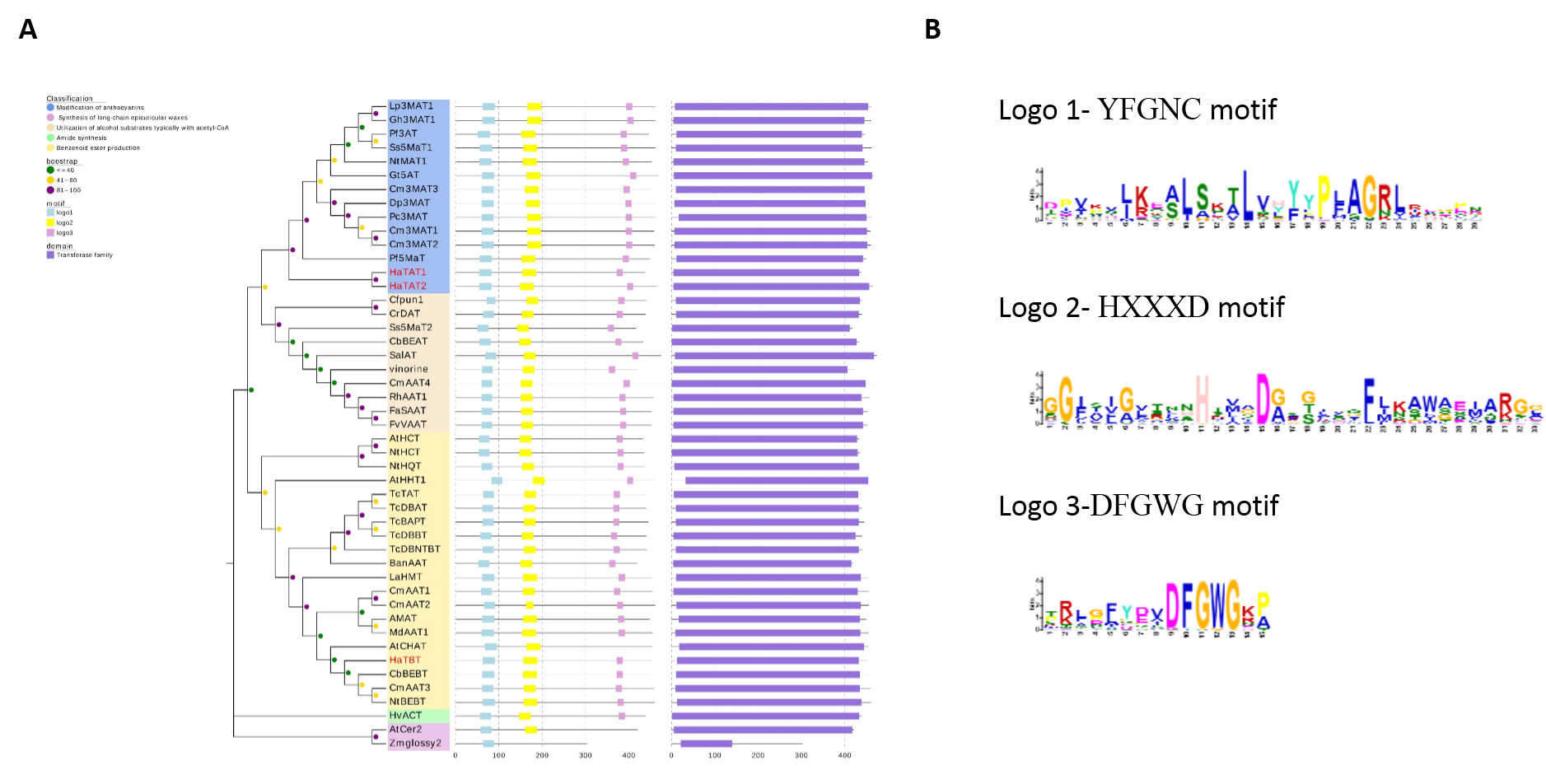
Meanwhile, the three candidate acyltransferase coding genes were selected to perform phylogenetic analyses with other functionally characterized BAHDs. HaTBT falls into branch that principally includes acyltransferases related to benzenoid ester production. HaTAT1 and HaTAT2 were aggregated into the same branch as other anthocyanin-modifying acetyl transferases (Figure S9A, Table S16). Domain analysis showed that the transferase family domain existed in these three candidate genes. And they all possess three amino acid motifs, HXXXD, YFGNC, and DFGWG (Figure S9B), which are highly conserved in BAHD enzymes [40].
Heterologous expression and functional characterization of the OSCs
Three candidate OSCs were functionally validated by expressing their ORFs in expression vector pESC-TRP. Recombinant plasmids were transferred into yeast (Saccharomyces cerevisiae strain WAT11) by lithium acetate conversion method. Protein expression of the recombinant yeast within 24 hours were investigated, which implied that the recombinant yeast pESC-TRP-HaOSC1 successfully expressed the protein with the molecular mass of about 86 kDa and the protein expression reached the highest level 8 hours after induction (Figure 6A).
Gal-induced transgenic yeast cells were also analyzed for the accumulation of triterpenoids metabolites. Yeast cells were extracted and metabolites were identified through gas chromatography-mass spectrometry (GC-MS) analysis. By comparing the retention time and mass spectra of those new products with authentic standards (a-amyrin, β-amyrin and lupeol), we found that HaOSC1-expressing transgenic yeast accumulated lupeol, which was not present in the control yeast cells transformed with empty vector (Figure 6B-C). The yeast expression experiments were repeated three times, always with very similar results. These results clearly manifested that HaOSC1 encoded a lupeol synthase, which could use 2, 3-oxidized squalene in yeast as substrate to synthesize the target product lupeol (Figure 6D).
3.5 Functional validation of the CYP450s
To determine the product specificities of HaCYP450s, ORF of four candidates was expressed in yeast under the control of the gal-inducible GAL10 promoter. Induced cells were further used for Western blot analysis, the fusion protein bands of HaCYPi3 of about 54 kDa were observed. The induction time was also investigated, same with HaOSC1, the protein expression reached the highest level at about 8 hours after induction (Figure 7A).
Based on the previous sequence analysis, HaCYPi3 was predicted to be a potential C-28 oxidase, catalyzing α-amyrin, β-amyrin and Lupeol substrates. Therefore, we firstly transferred pESC-HaCYPi3 and the above pESC-HaOSC1 recombinant plasmid into the same yeast for co-expression. The metabolites after induction were analyzed by GC-MS. Unfortunately, the target product betulinic acid was not detected, which suggested that HaCYPi3 failed to encode for a lupeol oxidase. Then, we co-expressed HaCYPi3 with α-/β-amyrin synthase IaAS previously identified by our research group from llex asprell. It is surprising that IaAS and HaCYPi3 co-expressing transgenic yeast accumulated new products compared with empty vector. Except for the corresponding substrate peaks, the retention time and mass spectra of one new product was consistent with oleanolic acid authentic standards (Figure 7 B-C). The results of three repeated experiments were consistent, which clearly demonstrated that HaCYPi3 encoded a β-amyrin oxidase, which could use β-amyrin catalyzed by IaAS in yeast as substrate to synthesize the target product oleanolic acid (Figure 7D). Furthermore, the subcellular localization of HaCYPi3 was observed, results displayed that it was localized in the cytoplasm (Figure 7E),which is consistent with reports that triterpenoids were synthesized in the cytoplasm.
{kind=link}
{kind=link}
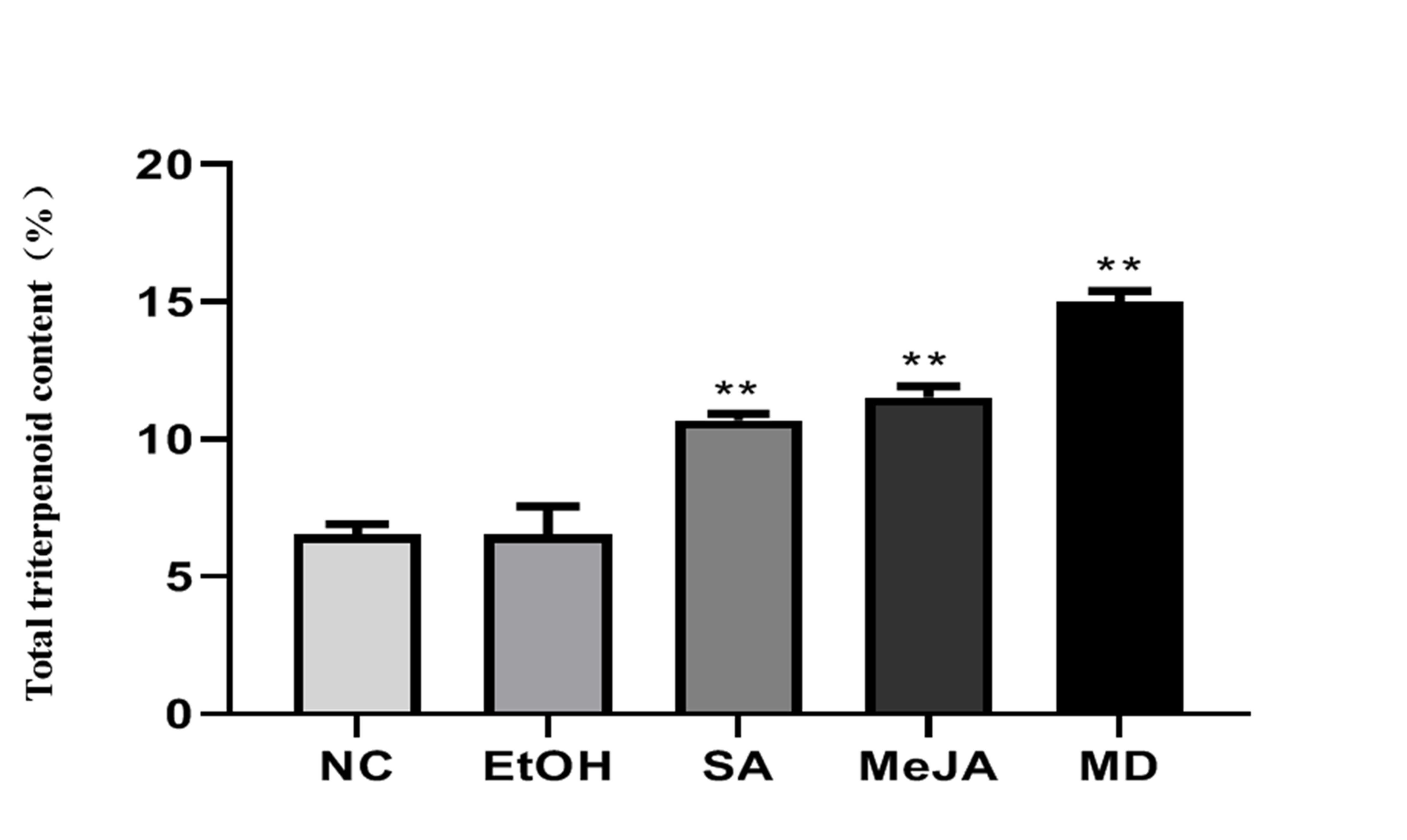{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}