Target protein identification
For the pivotal step of identifying target proteins, our approach began with extracting approved drug indications, particularly drug-indication combinations, from the ChEMBL database (version 29). ChEMBL is a comprehensive database cataloging bioactive molecules. It integrates extensive data on drug molecules, bioactivity, and genomics, thereby facilitating the development of new drugs with a genomic foundation.
Next, we systematically identified and selected drugs associated with these approved indications. We then utilized the ChEMBL API (see Supplementary Materials) to obtain the corresponding UniProt IDs of the target proteins. In instances where a drug was linked to multiple protein targets, we included each of these targets in our analysis to ensure a comprehensive evaluation. This methodological rigor allowed us to construct an inclusive dataset that was critical for the subsequent steps of our MR analysis.
GWAS summary data acquisition
Our approach to obtaining genome-wide association studies (GWAS) summary data for indications involved a consensus-driven process. This process was described by a review panel comprising three independent research participants. Each participant was subjected to a thorough search of the Integrative Epidemiology Unit (IEU) GWAS database (https://gwas.mrcieu.ac.uk/) to identify pertinent GWAS data aligned with the indications relevant to our study. To ensure robustness in our selection, we adopted a data validation strategy: if two or more participants agreed on the same GWAS dataset, it was directly incorporated into our analysis. In scenarios where there was a divergence of opinions, the panel engaged in detailed discussions to arrive at a consensus and make a final determination.
To broaden the scope and applicability of our study, we implemented strategic replacements for some GWAS datasets that were unavailable or incomplete. Our replacement methodologies included the following:
-
The disease subtypes were substituted for the overarching disease data when specific subtype data were absent.
-
Data from a representative subtype were used in place of the entire disease dataset.
-
Interchanging data between closely related subtypes of the same disease.
-
GWAS data are exchanged between diseases known to have causal relationships.
This meticulous process ensured comprehensive coverage and relevance of GWAS data, thereby augmenting the validity of our Mendelian randomization analysis.
Types of instrumental variables
The IVs employed in our MR analysis can be primarily divided into three distinct types:
Plasma pQTLs
We sourced plasma protein pQTL data from the research conducted by Benjamin B. Sun et al. [9]. This study comprehensively measured 2994 plasma proteins across 3301 participants of European ancestry using the advanced SomaLogic SomaScan platform. The data from this research are publicly available and provide a robust foundation for our plasma protein-focused analyses.
Tissue-specific eQTLs
Utilizing the resources of the Gene-Tissue Expression project (GTEx V.8), we obtained eQTL data for genes across 48 different tissues, including blood. This extensive dataset allowed us to incorporate a broad range of tissue-specific eQTLs as instrumental variables in our MR studies, enhancing the diversity and applicability of our analysis.
Brain pQTLs
We accessed brain tissue pQTL data from a study by Chloe Robins et al., which focused on genetic and proteomic analyses of 330 postmortem samples from the lateral prefrontal cortex of elderly individuals [10]. This study reported pQTL data for 7376 brain proteins, providing us with a comprehensive dataset for brain-specific MR analysis.
More information about the GWAS summary data of the target proteins can be found in Supplementary Table 1–3.
Composition of IV-Indication Pairs
To evaluate the impact of different frameworks on drug MR analysis, we generated sets of IV-indication pairs classified as either “true positive” or “true negative”.
True-positive target-indication pairs
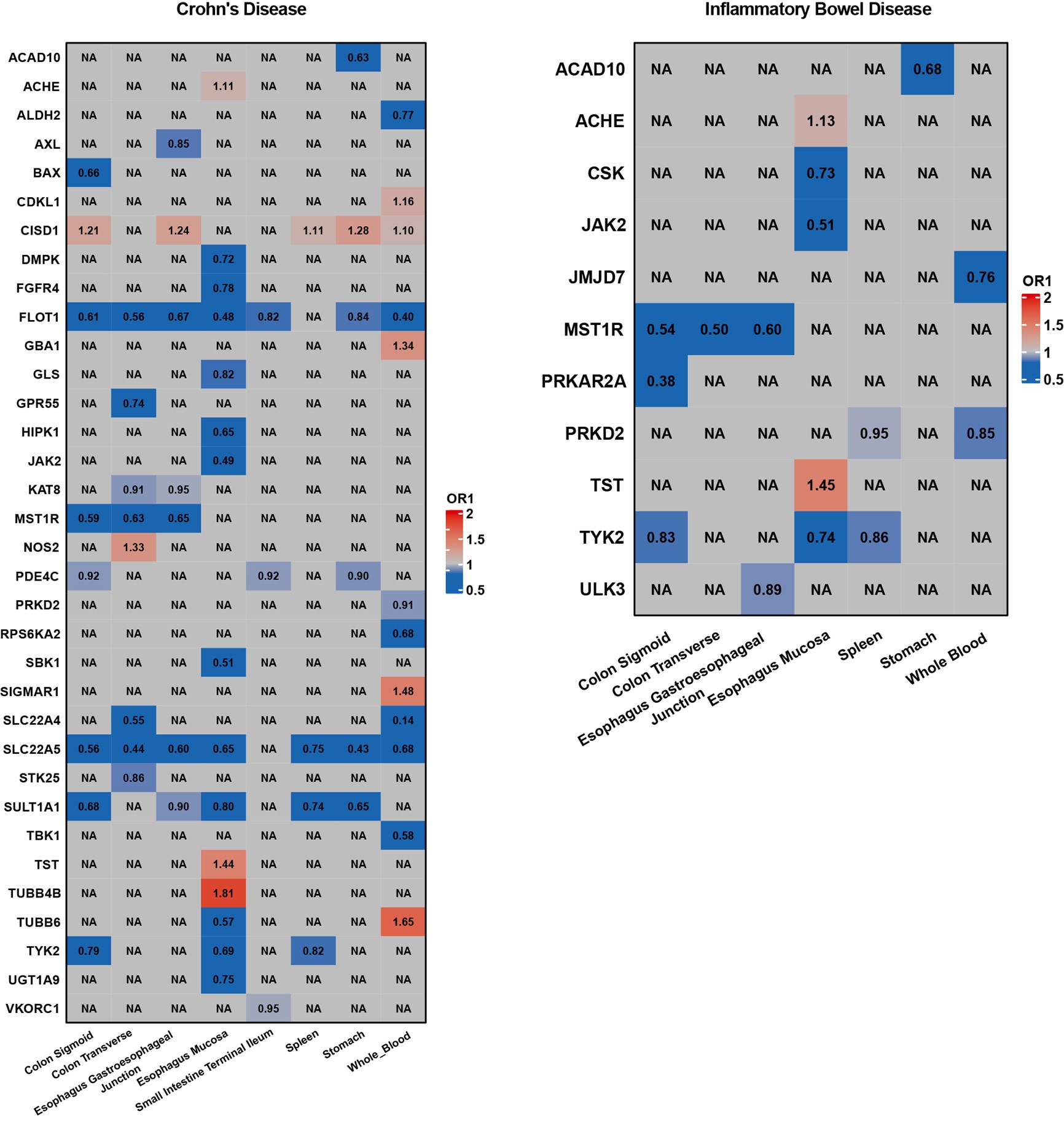
These pairs were derived from approved drug indications, with drug targets as the exposure and the indications as the outcome. Based on the instrumental variables used, we constructed five distinct sets of true positive pairs. These include plasma protein pQTLs for all related indications, plasma pQTLs for blood-related indications, blood eQTLs for blood-related indications, tissue-specific eQTLs for all related indications, and brain pQTLs for brain indications. The underlying assumption for these pairs is the existence of a causal relationship between the drug targets and indications, as evidenced by clinical trial efficacy, thus suggesting potential positive MR results. This method allowed us to assess the true positive rate and false negative rate under various conditions.
True-negative target-indication pairs
These pairs were used to evaluate the ability to correctly identify negatives. We merged eQTLs from different tissues with plasma pQTLs to create a comprehensive SNP pool. For each indication in the true-positive target-indication pairs, the same number of SNPs were randomly sampled from the SNP pool. Since these pairs are constructed with hypothetical target proteins without any known causal relationship to the indications, the anticipated MR results for these pairs are negative.
Determining the optimal LD r2 threshold
In MR analysis, removing redundant SNPs by LD serves as a balance between the bias induced by correlated SNPs and the instrumental power. To find an optimal LD r2 threshold, we applied various r2 values (0.001, 0.01, 0.2, 0.3, 0.4, 0.5, and 0.6) within a 500 kb clumping window to remove LD in the tissue-specific eQTLs of true-positive IV-indication pairs. Next, we performed MR analysis across different models to observe the effect of varying LD r2 values on the true positive rate. The optimal LD r2 value was ascertained based on these analyses. This value represents the threshold at which the true positive rate is maximized while avoiding the introduction of bias due to excessive LD among the IVs.
Determining the optimal P value threshold
Next, we clumped the instrument variables in the positive datasets of tissue-specific eQTLs using the optimal LD r2 obtained in the previous steps and performed MR analysis in both the positive and negative datasets. FDR correction was then applied to the P values in the obtained results. Subsequently, we screened each model's original and FDR-corrected P values using four commonly used filtering thresholds of 0.1, 0.05, 0.01, and 0.001 to obtain significant results and constructed a confusion matrix of each model under each P value threshold to determine the optimal P value threshold.
Effect of instrumental variables and model selection on the effectiveness of drug MR
We utilized the entire dataset, including the true positive and true negative datasets, to calculate causal relationships between exposure and outcome using five different algorithms. Subsequently, we constructed confusion matrices to evaluate the performance across different datasets and determine which algorithm model exhibited the highest true positive rate. This analysis aimed to assess the most effective dataset and identify the algorithm with the highest true positive rate.
Mendelian randomization analysis
Using the R packages TwoSampleMR (version 0.5.6) and MRPRESSO (version 1.0), we conducted MR analysis by applying five models: the Wald ratio, inverse variance weighted (IVW), MR‒Egger, weighted median, and MRPRESSO models. When heterogeneity existed, we used the results from the IVW random effects model as the final effect size. Otherwise, we used the fixed effect model. To ensure the validity of MR analysis, the selected instrumental variables (IVs) should meet the following three critical principles: (1) the IVs should be strongly associated with the gene expression level or protein expression level (P < 5×10− 8). (2) There should be no association between the IVs and the outcome variables (i.e., the indications). (3) There should be no association between the IVs and confounding factors.
In this study, \({\widehat{\beta }}_{j}\) represents the causal effect of exposure on the outcome, \({\widehat{\gamma }}_{j}\) represents the strength of the association between genetic variants and exposure, and \({\widehat{\gamma }}_{j}\) is the regression coefficient of the outcome on each genetic variant. For cases with multiple instrumental variables, we employed five models for MR calculation: the Wald ratio, IVW, MR‒Egger, weighted median, and MRPRESSO models[11–13].
Statistical analysis
We defined a drug indication (i.e., a combination of a disease and a drug) as a sample and performed statistics on the results of five models: Wald ratio, IVW, MR‒Egger, weighted median, and MRPRESSO. The total sample size N for each model is the total number of drug indications in the results. Then, we screened significant results using a selected P value threshold and excluded results that were driven by pleiotropy. In the true positive datasets, the direction of the effect of the target proteins on the significant results should also be the same as that reported in the ChEMBL database. For drug indications composed of multitarget drugs, we considered the sample’s result to be significant if the result of any target was significant. We then counted the number of significant samples for each model and obtained the positive sample size n for the model. The true positive rate and false positive rate of drug prediction using the model can be calculated from the formula n/N in the positive and negative datasets, respectively.
In MR analysis, we used Cochrane's Q test to assess heterogeneity and MR‒Egger regression to assess pleiotropy. Then, we calculate the F statistic, which measures the effectiveness of the instrumental variables[14]. It is important to note that the method used to calculate r2 is only an approximation due to the lack of sample genotype data.
In this study, we used positive and negative datasets under the same conditions and calculated the following metrics:
The accuracy represents the proportion of correct predictions out of the total samples and is expressed as follows:
$$\text{A}\text{c}\text{c}\text{u}\text{r}\text{a}\text{c}\text{y}=\frac{TP+TN}{TP+TN+FP+FN}$$
Precision refers to the probability of a positive sample actually being positive, given that it has been predicted to be positive. It is expressed as:
$$\text{P}\text{r}\text{e}\text{c}\text{i}\text{s}\text{i}\text{o}\text{n}=\frac{TP}{TP+FP}$$
Recall is a metric that considers the original sample. Its meaning is the probability of an actual positive sample being predicted as positive. The expression is as follows:
$$\text{R}\text{e}\text{c}\text{a}\text{l}\text{l}=\frac{TP}{TP+FN}$$
The F1-score is the harmonic mean of precision and recall, which considers both precision and recall to reach the highest level simultaneously and achieve a balance.
$$\text{F}1-\text{S}\text{c}\text{o}\text{r}\text{e}=\frac{2*\text{P}\text{r}\text{e}\text{c}\text{i}\text{s}\text{i}\text{o}\text{n}\text{*}\text{R}\text{e}\text{c}\text{a}\text{l}\text{l}}{\text{P}\text{r}\text{e}\text{c}\text{i}\text{s}\text{i}\text{o}\text{n}+\text{R}\text{e}\text{c}\text{a}\text{l}\text{l}}$$
{kind=link}