PIP5K expression increased along with autophagy and antioxidant markers in HCC patient samples and cell lines
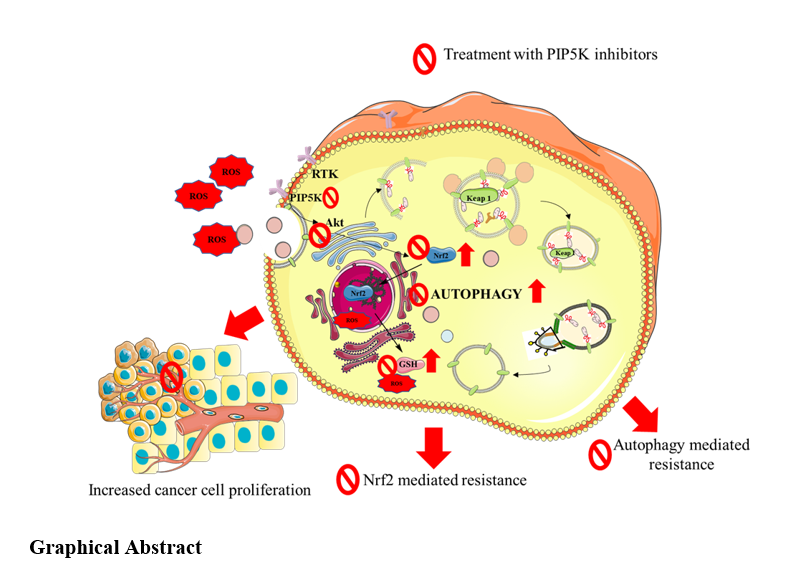
To investigate the potential link between the PIP5K-autophagy and Nrf2 and to explore the role of PIP5K in the progression of hepatic cancer, we estimated the expression of PIP5K1A, PIP5K1B, Beclin-1, and Nrf2 in 36 human HCC patient samples. We compared the expression of these markers in various cancer grades (G1-G3) with that in normal adjacent control tissues (UAT). We observed a grade-dependent increase in PIP5K1A, PIP5K1B, Beclin-1, and Nrf2 expression. PIP5K1A and PIP5K1B expression levels were elevated in all three grades of cancer (G1, G3 * (p < 0.05), and G2 ** (p < 0.01)). We observed the highest expression of the PIP5K isoforms in G2. A similar trend was observed for Nrf2 expression (G1 * (p < 0.05) and G2 **(p < 0.01)). However, in the case of autophagy marker Beclin-1, we observed a steady grade-dependent increase in expression, with the highest expression observed in G3 (**, p < 0.01), when compared to normal adjacent control tissues (Fig. 1A, B, C, and D).
A similar trend of positive correlation between PIP5K isoforms, Beclin-1, and Nrf2 was observed when we estimated the basal expression levels in various HCC cell lines such as PRF5, SNU387, Skhep-1, and HepG2 (Fig. 1E and F). The expression levels of PIP5K isoforms, Beclin-1, and Nrf2 were low in PRF5 and SNU-387 cell lines, whereas the expression of PIP5K isoforms (PIP5K1A and B) was higher in HepG2 cells (** p < 0.01 for both PIP5K1A and B) compared to PRF5 cells. Correspondingly, Beclin-1 and Nrf2 (* p < 0.05, Beclin-1 and *** p < 0.001 Nrf2 compared to PRF5) levels were elevated in HepG2 cells. Keap-1 is a degrading protein responsible for preventing nuclear translocation of Nrf2. Basal Keap-1 levels were low in HepG2 cells and highest in Skhep-1 (*** p < 0.001 compared to PRF5). The Nrf2 dependant-antioxidant enzymes HO-1 (** p < 0.01), SOD2 (* p < 0.05), and AKR1B10 (*** p < 0.001) were also elevated in HepG2 cells compared to the PRF5 cell line (Fig. 1C and D). The positive correlation between proliferative markers (PIP5K isoforms) and adaptive markers (such as Beclin-1, Nrf2, HO-1, and SOD2) suggests that these markers work together to sustain proliferation and energy supply and counter ROS. Because HepG2 cells express high basal levels of PIP5K isoforms, autophagy, and Nrf2-mediated antioxidant markers, HepG2 cells were selected for further experiments.
PIP5K resists ROS-mediated autophagic cell death
ROS govern various cell phases such as proliferation, adaptation, and death by modulating the expression levels of lipid kinases. Thus, to establish the role of ROS in the modulation of PIP5K and the autophagy-Nrf2 adaptive axis, we treated HepG2 cells with increasing concentrations of H2O2 (6.25, 12.5, 25, 50, 100, and 200 µM) and explored its effect on cell viability, mitochondrial superoxide (ROS), lysosomal turnover (autophagy flux), expression of PI3K-Akt, autophagy, antioxidant, and apoptosis markers (Fig. 2). To estimate the concentrations of H2O2 (ROS) responsible for proliferation, stasis, and death, HepG2 cells were treated with increasing concentrations of H2O2 and subjected to the MTT assay. We observed a dose-dependent increase in cell proliferation at lower doses of 6.25 (###, p < 0.001 vs. untreated cells) and 12.5 µM (##, p < 0.01, vs. untreated cells). Reactive Oxygen Species (ROS) generated aided growth as an autophagy-mediated antioxidant pathway countered the ROS, and the energy generated in turn drove the cell growth observed in increased cell proliferation at lower H2O2 concentrations. This was followed by an adaptive phase, as observed at 25 and 50 µM, where cells that underwent autophagy induction could not generate sufficient energy and nutrients for cell division, as indicated by reduced cellular proliferation, high ROS levels, and lysosome turnover. This phase was followed by autophagic cell death at higher H2O2 concentrations (100 and 200 µM), where elevated levels of ROS triggered autophagic cell death. Such dose-dependent behavior with increasing H2O2 concentration was not observed in the serum-containing complete media (Fig. 2A). The MitoSOX assay showed that mitochondrial superoxide concentration following H2O2 exposure increased with increasing H2O2 concentration. Serum starvation increased ROS levels compared to cells grown in complete media [*, p < 0.05, compared to complete media (CM)]. ROS levels remained fairly unchanged even at increased H2O2 concentrations (50 µM). However, at cytotoxic concentrations of 100 and 200 µM, mitochondrial superoxide levels further increased compared to those in the starvation control. The increase in mitochondrial superoxide concentration was ## p < 0.01 and ### p < 0.001, respectively, when compared to the serum starvation control (Fig. 2C). Concurrently, when we measured lysosomal turnover indicative of autophagic flux increased following starvation (*, p < 0.05, compared to CM) following exposure to H2O2 in serum-starved cells, we observed a dose-dependent increase in lysosomal turnover compared to CM (Fig. 2B). H2O2 at 200 µM induced more autophagic-lysosomal turnover than serum starvation (###, p < 0.001 compared to serum starvation), resulting in autophagic cell death (Fig. 2B). Taken together, the results of cellular proliferation, MitoSOX, lysotracker staining, and mitochondrial superoxide (ROS) indicated that autophagy was activated. The elevated autophagy resisted an increase in mitochondrial superoxide at mild concentrations (6.25 and 12.5 µM), which induced cellular proliferation at moderate concentrations (25 and 50 µM) and promoted cellular stasis. However, cytotoxic concentrations of 100 and 200 µM enhanced ROS to cytotoxic levels, thereby overactivating autophagy and leading to autophagic cell death (Fig. 2B). Dual PIP5K1B and lysotracker staining indicated that both PIP5K1B expression and lysosomal turnover were elevated by 12.5 µM H2O2 (Fig. 2D). Western blotting showed that gradient-increasing H2O2 exposure affected the expression of PIP5K isoforms (PIP5K1A and B), Nrf2, SOD2, and HO-1. Serum starvation alone increased PIP5K1A, PIP5K1B, and SOD2 expression compared with CM (*, p < 0.05, p < 0.01). There was a consistent increase in the expression of PIP5K1A (** p < 0.001 vs. CM and #p < 0.05 vs. starvation), PIP5K1B (**p < 0.01 vs. CM and #p < 0.05 vs. starvation), Beclin-1 (**p < 0.01), and SOD2 (**p < 0.01 vs. CM and #p < 0.05 vs. starvation) as the concentration of H2O2 increased to cytostatic levels at 50 µM (Fig. 2E and F). However, Nrf2, HO-1, Bax, and BCl2 expression remain unaffected by treatment with 50 µM H2O2. However, at a cytotoxic concentration, especially at 200 µM, the expression of PIP5K1A, PIP5K1B, Nrf2 (## p < 0.01 vs. starvation), SOD2 (# p < 0.01 vs. starvation), HO-1 (###, p < 0.001 vs. starvation), and Beclin-1 (**, p < 0.001 vs. CM and #, p < 0.05, vs. starvation) were decreased. SRC (##, p < 0.001 vs. starvation), a marker responsible for the relocation of PIP5K from membrane-based signaling to cytosolic and apoptotic proteins Bax/BCl2 ratio increased following 200 µM H2O2 exposure (Fig. 2D and E). These results indicate that cells increased PIP5K expression to maintain both PI3K-Akt proliferative and autophagy signaling, upregulating Nrf2 mediated antioxidant defence markers, and increasing SRC indicated a shift from membrane-based proliferative PI3K-Akt to cytosolic autophagy signaling (9, 10). However, at a cytotoxic concentration of 200 µM, the expression of PIP5K isoforms Nrf2, SOD2, and HO-1 decreased, while that of SRC and Beclin-1 increased. This indicates that cells exposed to high H2O2 (ROS) concentrations could not modulate ROS even by upregulating autophagy and the Nrf2 mediated antioxidant defence pathway, thus resulting in the overactivation of autophagy, causing autophagic cell death (Fig. 2D and E). Thus, to mimic tumor microenvironment, with elevated ROS, limited nutrients, and cells in a rapidly proliferative phase, HepG2 cells exposed to mild ROS (12.5 µM H2O2) along with serum-starvation condition was chosen as it exhibited consistent results with increased cellular proliferation, autophagy, and antioxidant defence mechanism.
Identification of new lead PIP5K inhibitor NG-TZ-17
Under lead optimization, biologically active derivatives of IITZ01 were screened for PIP5K1B inhibition, and NG-TZ-17 and NG-TZ-20 also exhibited PIP5K1B inhibition of 73% and 57%, respectively, at 1 µM (Fig. 3A). The PIP5K1B enzyme inhibition IC50 values of the identified inhibitors NG-TZ-17, IITZ01, and NG-TZ-20 were 239, 281, and 691 nM, respectively (Fig. 3B). Structure-activity relationships revealed that compounds substituted with phenyl groups (IITZ01, NG-TZ-20) showed decreased activity compared to the simple phenyl group (NG-TZ-17).
Triazine PIP5K inhibitors were superior to standard PIP5K1A and autophagy inhibitors in inhibiting proliferation and autophagy
We explored the efficacy of investigational triazine PIP5K inhibitors IITZ01, NG-TZ-17, and NG-TZ-20, along with standards PIP5K1A and autophagy inhibitors ISA-2011B and Chloroquine (CQ) in hepatic cancer cell lines such as HepG2, SNU-387, Skhep-1, and PRF5 (Fig. 3C). We observed that IC50 of NG-TZ-17, IITZ01, NG-TZ-20, ISA-2011B and chloroquine (CQ) in Hepatic cancer were 0.94, 1.63, 5.66, 7.76, and 67.10 µM, respectively in HepG2. IC50 of NG-TZ-17, IITZ01, NG-TZ-20, ISA-2011B and chloroquine (CQ) were 6.48, 1.87, 2.59, 5.59, and 20.69 µM, respectively in SNU-387 cell line. In Skep-1, the concentrations of NG-TZ-17, IITZ01, NG-TZ-20, ISA-2011B, and chloroquine (CQ) they were 2.54, 3.21, 6.47, 8.20, and 54.75 µM, respectively. Finally, in PRF5, IC50 of NG-TZ-17, IITZ01, NG-TZ-20, ISA-2011B, and chloroquine (CQ) were 1.70, 2.80, 4.07, 4.21, and 11.13 µM, respectively. Overall, we observed that the triazine PIP5K1B inhibitors NG-TZ-17 and IITZ01 showed better inhibition of proliferation than standard ISA-2011B (PIP5K1A inhibitor) and chloroquine (autophagy inhibitor). NG-TZ-20 proliferation inhibitory activity was equipotent to that of standard ISA-2011B and superior to that of chloroquine (CQ) (Fig. 3C). In order to explore the effect of triazine PIP5K inhibitors on autophagy, we treated HepG2 cells with two doses of NG-TZ-17 [0.5 µM (17 L) and 1 µM (17H)], IITZ01 [0.5 µM (IITZ01 L) and 1 µM (IITZ01 H)], and NG-TZ-20 [(2.83 µM (20 L) and 5.66 µM (20H)]. Acridine orange-red staining indicated that NG-TZ-17, IITZ01, and NG-TZ-20 deacidified acidic vesicles inhibited autophagy. The treatment resulted in a decrease in acidic vesicles NG-TZ-17 (##, p < 0.01, and ###, 0.001 for NG-TZ-17 at low and high concentrations, respectively, vs. starvation control), and both concentrations of NG-TZ-20 and IITZ01 significantly reduced acidic vesicles (###, p < 0.001 vs. starvation control) (Fig. 3D). To determine the stage-specific inhibition of autophagy, we transfected HepG2 cells with the LC3B-GFP-RFP peptide, which is incorporated into autophagosomes when autophagy is stimulated. Treatment with the triazine PIP5K inhibitors NG-TZ-17 and IITZ01 resulted in an increase in both GFP-LC3B and RFP-LC3B puncta, indicating inhibition of autophagy at the autolysosomal step (Fig. 3E and F). Furthermore, we performed PIP5K1B immunostaining and lysotracker dual staining (Fig. 4). PIP5K inhibitors reduced PIP5K1B expression and lysosomes at high doses of NG-TZ-17, IITZ01, and ISA-2011B. The chloroquine (CQ) standard successfully reduced lysosomal expression (Fig. 4). However, chloroquine treatment did not affect the expression of PIP5K1B. These results indicate that the inhibition of autophagy following treatment with triazine derivatives was due to a decrease in PIP5K1B expression.
Treatment with PIP5K inhibitor sensitized HepG2 to the mild concentration of H2O2
Tumor cells modulate ROS levels to maintain high proliferation (11). Tumor cells modulate the ROS switch between proliferation and adaptive pathways (11). Thus, to mimic the tumor microenvironment with elevated ROS levels, we stimulated HepG2 cells with a low concentration of H2O2. We exposed the starved HepG2 cells to 12.5 µM H2O2. The H2O2 exposed cells were then treated with two doses of NG-TZ-17 [(0.47 (17 L) and 0.9 µM (17H)] and IITZ01 [0.5 (IITZ01 L) and 1 µM (IITZ01 H)] along with standards 7.76 µM of ISA-2011B (PIP5K1A inhibitor), 67.10 µM of Chloroquine (autophagy inhibitor), and 5 µM ML-385 (Nrf2 inhibitor). We estimated the levels of mitochondrial superoxide, PIP5K1B, lysosomal turnover, and expression of PI3K-Akt, autophagy, and Nrf2 antioxidant defense markers following treatment with inhibitors.
The MitoSOX staining results revealed that mitochondrial superoxide levels were increased when stimulated with 12.5 µM of H2O2 (*, p < 0.05 vs. CM). Treatment with PIP5K inhibitors NG-TZ-17, IITZ01, and ISA-2011B, along with autophagy (CQ) and Nrf2 (ML-385) inhibitors, increased mitochondrial superoxide levels (Fig. 5A and B). The effects of PIP5K inhibitor treatment were similar to those of Nrf2 (ML-385) and autophagy inhibitors (chloroquine). High doses of NG-TZ-20 and IITZ01 significantly increased mitochondrial superoxide levels (#, p < 0.05) compared to starved cells exposed to H2O2 (12.5µM) (Fig. 5A and B). Further, to determine the molecular effects of treatment with PIP5K inhibitor on H2O2 exposed cells, we explored the effects of treatment on the PI3K-Akt, autophagy, and Nrf2 pathways. H2O2 (12.5 µM) exposure increased p-mTOR (**, p < 0.01), Nrf2 (*, p < 0.05), PIP5K1B (***, p < 0.001), PIP5K1A (***, p < 0.001), Beclin-1 (*, p < 0.05), and SRC (*, p < 0.05) compared to cells grown in complete media (Fig. 4E and F). Treatment resulted in decrease in mTOR (#, p < 0.05, for both concentrations of IITZ01 H and ISA-2011B), Nrf2 (#, p < 0.05for17 H, IITZ01 H, ISA-2011B, CQ, and ###, p < 0.001 for ML-385), PIP5K1B (###, p < 0.001 for 17H and ##, p < 0.01 for both concentrations ofIITZ01), PIP5K1A (##, p < 0.01 for 17H, #, p < 0.05 for both concentrations of IITZ01, and ###, p < 0.001 for ISA-2011B), p-AKT (#, p < 0.05 for 17H, both concentrations of IITZ01 and ISA-2011B), HO-1 (#, p < 0.05 for 17H, and both concentrations of IITZ01), SOD2 (#, p < 0.05 for 17H, for both concentrations of IITZ01, ISA-2011B, and CQ), Beclin-1 (#, p < 0.5 for 17H and IITZ01 L), Bax/BCL2 ratio(##, p < 0.01 for 17 L, ###, p < 0.001, for 17H and #, p < 0.05 for IITZ01 H) and no significant changes in SRC expression levels compared to 12.5 µM H2O2 exposed group (Fig. 5B and C). Furthermore, to validate the results of molecular analysis, we performed PIP5K1B and lysotracker dual staining, where we observed that Chloroquine, and ML-385 did not have any effect on PIP5K1B expression, although chloroquine was able to inhibit autophagy, as indicated by an increase in Lysotracker fluorescence. Treatment with PIP5K inhibitors, both investigational NG-TZ-17, NG-TZ-20, and IITZ01, and standard ISA-2011B, resulted in a decrease in PIP5K1B and lysosome expression (Fig. 6). These results indicate that NG-TZ-17, IITZ01, and ISA-2011B (PIP5K inhibitors) are superior to the standard autophagy (chloroquine (CQ)) and Nrf2 (ML-385) inhibitors. Furthermore, the triazine lysosomotropic PIP5K inhibitors NG-TZ-17 and IITZ01 were superior to the standard ISA-2011B (PIP5K1A) inhibitor in sensitizing HepG2 cells to a mild concentration of H2O2. This was due to the dual inhibition of the PI3K-AKT and autophagy pathways.
Inhibition of proliferative, autophagy and Nrf2 pathways by PIP5K inhibitors reduced tumor burden in HepG2-induced hepatic cancer in SCID mice
GFP-tagged HepG2 cells were injected subcutaneously into the flank region of SCID mice. After the tumors attained 200 mm3volume, mice were treated with PIP5K inhibitors (NG-TZ-17 and IITZ01) and sorafenib for 7 days (Fig. 7A). Tumor-induced mice were treated with dailyNG-TZ-17 (50 mg/kg), IITZ01 (50 mg/kg), or sorafenib (60 mg/kg) for 10 days. The treatment resulted in a decrease in tumor burden, weight, and volume (Fig. 7B-E). The reduction in tumor burden was equipotent to sorafenib (***, p < 0.001 for NG-TZ-17, IITZ01, and Sorafenib when compared to tumor-bearing mice) (Fig. 7E). Treatment with PIP5K inhibitors and sorafenib increased the overall survival of the tumor-induced mice compared to that of the control tumors (Fig. 7C). Western blot examination of isolated liver carcinoma tumors revealed that treatment with PIP5K inhibitors (NG-TZ-17 and IITZ01) reduced the proliferative pathway, as evidenced by a decrease in PIP5K isoforms such as PIP5K1A (**, p < 0.01 for NG-TZ-17, IITZ01, and * p < 0.05, for sorafenib), PIP5K1B (***, p < 0.001 for NG-TZ-17 and IITZ01), and p-Akt (*, p < 0.05 for NG-TZ-17, Sorafenib and **, p < 0.01 for IITZ01). PIP5K treatment also inhibited autophagy, as indicated by a reduction in Beclin-1 levels (***, p < 0.001 for NG-TZ-17 and *, p < 0.05 for IITZ01). Inhibition of the proliferative and autophagy pathways resulted in inhibition of Nrf2 dependant antioxidant pathway, as indicated by the depletion Nrf2 expression (**, p < 0.01 for NG-TZ-17 and IITZ01). Treatment with both PIP5K inhibitors (NG-TZ-17 and IITZ01) and sorafenib induced apoptosis, as indicated by an increase in Bax/BCL-2 ratio expression (**, p < 0.01 for NG-TZ-17, IITZ01, and sorafenib), when compared to the tumor-bearing group (Fig. 7F and G).
{kind=link}