3.1 Query types.
SLiMAn can be interrogated in two ways depending on the data available. Either a given interactome has been obtained (or selected from any source of data) and the corresponding list of proteins can be directly submitted to the webserver or the submitted entry corresponds to only one unique protein. In the latter case, SLiMAn interrogates BioGRID and/or IntAct to retrieve a (meta-)interactome. This constitutes a list of putative interactants to be submitted directly and analyzed using the very same webserver. Once the query list is submitted, the same workflow applies to the data regardless of their origin (Figure 1). In the case an original interactome is submitted, the data from BioGRID and/or IntAct can serve as a validation, as illustrated previously [26] and further in this protocol.
To illustrate the different properties of SLiMAn2 for interactomic data analysis, a focused proteomic study on tankyrase 1 and 2 published by Li and colleagues in 2017, name hereafter the TNKS1/2 interactome is proposed [30]. The information extracted from this analysis is compared to the one resulting from performing a parallel analysis of the meta-interactome extracted from BioGRID and IntAct for the same two tankyrases. The goal is to define the molecular features of the different PPIs present in the interactomes from distinct sources and to reveal potential SLiM-based interactions involved in different functional networks. We describe below the step-by-step method used to partially decipher a SLiM-based protein network.
3.1.1. Querying a novel interactome
1. Collect identifiers (or accession codes) from Uniprot for all the partners of the TNKS1/2 interactome.
2. Start a new project on the webserver by submitting the list of names – separated by commas – on, the front page (Figure 2A). A project name can be input in the dedicated window.
3. Press the button “Find Interactions” just below and SLiMAn will start its analysis (see below 3.2).
3.1.2. Quick start from a given protein.
Alternatively:
- Submit the Uniprot names for TNKS1 or 2 (TNKS1_HUMAN or TNKS2_HUMAN, respectively) to the menu “PPI Extension” on the SLiMAn front page (Figure 2B). In this case, SLiMAn queries BioGRID and IntAct that contain curated protein interactomic data for most model organism species.
- Press “Find Interactants” and SLiMAn-2 will retrieve all the interactomic data from these two databases (see 3.1.3).
3.1.3 Filtering putative interactants
From a given UniProt protein name (or code), SLiMAn rapidly extracts the putative partners listed in BioGRID and/or IntAct. A resulting webpage interactively enlists, by default, the proteins associated with the query in any of those two databases. This list can range from very few to up to a thousand of protein partners. SLiMAn can manage more than one hundred of interactors but not a thousand in the current version. However, it is likely too difficult to survey too many partners at once. Hence, the number of interactants to study, can be filtered using the «Parameters» section shown just above the protein list. Data from only one database can be used instead or, on the contrary, one can focus on the intersection of the two databases, BioGRID and IntAct, for higher confidence. Note that there is a significant overlap between the two databases and in that case, this redundancy cannot be seen as a cross-validation. Otherwise, as the data from BioGRID are separated in low- and high-throughput classes while IntAct data are split in general data and those from HuRI (http://www.interactome-atlas.org/; [10]), one can reduce further and tune the query list before submission. For example, only low-throughput data from the BioGRID database can be selected or, instead, only HuRI proteomic data (direct PPIs) within IntAct data. For higher confidence, filtering in interactants detected by both low- and high-throughput methods and present in both databases, is a good choice, although at the expense of the number of partners. Each time, SLiMAn updates the list of proteins to survey. One can hoover the mouse on the query name to read the number of interactants enlisted.
Once parameters are chosen, press the button “Quick launch” to launch SLiMAn analysis.
3.2 Main outputs.
Whatever the type of data (provided list of protein or meta-data extracted for one given protein), SLiMAn will search for ELM motifs as well as the corresponding PFam domains (see Note 1) within the submitted protein sequences. To filter only relevant information, orphan or unpaired ELM motifs and PFam domains are discarded. This dramatically reduces the list of ELMs amenable to analysis, in contrast to a direct interrogation of the ELM database. The motifs and domains filtered in, are shown on the main table for further interactive analysis. This table highlights all the putative pairing between an ELM motif and the corresponding domain. Numerous parameters can be used to filter in or out, more or less protein pairs as illustrated below.
3.2.1. General view.
In its upper part, the main result page recapitulates the query parameters (query name, number of partners, …). After an automatic setting of the filters (see Note 2), it tabulates the number of ELM motifs, PFam domains and number of proteins containing (or not) such motifs and/or domains (Figure 3). It also provides links to useful outputs for subsequent studies.
In the central part of the webpage, eight modules of different filters are present with the corresponding buttons, cursors or digits for selection (Figure 4). The “ELM”, “Disorder”, “HSM” and “PSP+” filters allow to manage structural and molecular parameters of PPis (Figure 4A). The second part of the filter panel is dedicated to the selection (“PPI database”, “SLiMan” (level of confidence),”visual”) and display forms (“visual”) of PPis (Figure 4B). Several examples of the roles of this interface in interactive analysis of an interactome is devellop in more details below (see 3.3).
In the lower part, a table is provided in which the ELM-PFam pairing computed by SLiMAn are highlighted. In the top row, PFam domains are listed associated with the UniProt identifier of the proteins containing them. In the left column, SLiMs corresponding to ELM entries, are listed in association with the UniProt identifier of the proteins containing them. Molecular features for each SLiM are detailed, namely, its ELM class, the corresponding regular expression and sequence motif as well as its location in the studied protein. If a PTM is annotated in the motif, as extracted from PhosphoSitePlus (herein PSP+; [31]), it is also highlighted (see below for more details).
For each ELM-PFam pair highlighted by SLiMAn, a box is drawn and colored according to the corresponding confidence level. It contains three links and a check box. The latter can be used to select a pairing and collect the corresponding validated pairs for subsequent visualization in a table or a Cytoscape network (see below) [32]. The “Hit” link (upper right panel) opens up a pop-up window recapitulating all the parameters computed for the match (see Figure 5 and following sections for further details). The lower left box is a link to an alignment module and the possible launch of comparative modeling of the putative complex that the ELM motif and the matched domain could form (see 3.4). When actual modeling within the framework of SLiMAn is performed, a new link is created in the lower right box of the main result page. These additional steps may help assessing the likelihood of a pairing.
Finally, below each column in the main table, a consensus sequence is computed and highlighted using LogoJS [33] for each ELM motif type paired with a given PFam domain. A selection button enables switching from one ELM type to another (e.g.: LIG_SH3_1 to LIG_SH3_9).
Modifying thresholds for the various parameters triggers a new computation resulting in a new table of pairings and new logos.
3.2.2. Specific information for each ELM-PFam pair.
In each ELM/PFam pair box within the main table, pressing the “Hit” button grants direct access to information regarding a given putative interaction in a dedicated pop-up window (Figure 4). This window details the ELM entry (motif name and E-value, sequence boundaries and whether or not it is part of the validated instance according to ELM), the experimental data extracted from BioGRID and IntAct databases (indicating the number of hits and providing links to the associated publications in PubMed), key biophysical parameters from IUPred2A [34] and AlphaFold [35] as well as the pairing likelihood score computed by HSM [36].
The IUPred2A section highlights the different scores (ANCHOR, IUPred local and global disorder or domain scores) for protein disorder predictions [34]. SLiMAn2 also provides the pLDDT score from pre-computed models stored in the AlphaFold database (AFDB) [29]. This score estimates the local accuracy, and was observed to nicely correlate to local disorder computed by IUPred. HSM is a dedicated predictor of interaction likelihood (from 0 to 1) for 6 types of recognition domains (PDZ, SH2, SH3, WW, WH1 and PTB) [36].
The number of experiments corresponding to a given pair of proteins within BioGRID and IntAct are split in categories (e.g.: Low vs High throughput methods for BioGRID data, as discuss above). Select “toggle details” to see the precise type of experiments used, the date of deposit in the database (for IntAct data), a reference in PubMed as well as a link to the associated publication (as extracted from BioGRID and IntAct information). Additional technical information from IntAct and HuRI are also displayed (see Note 3).
In the upper-right corner of the pop-up window, “PubMed queries” links are provided to search PubMed for publications describing each protein as well as those associating the two proteins of that pair (Figure 6). The latter corresponds to a search in PubMed combining all the alternative identifiers, accession and gene names for each protein in the pair, using logical operators. It enables quick access to related research articles found in literature and can compensate the lack of deposition in PPI repositories.
3.3 Hands on SLiMAn interactivity
The main page of results shows a subset of putative pairings extracted from the query list with a set of parameters tuned automatically (Figure 5). This works well with most queries, highlighting some ELM motifs but hiding many others due to current parameters such as, a too stringent (= small) E-value, or restrictive disorder parameters. The interactive fine-tuning of parameters supports the identification of promising pairings and the detection of direct interactions through a given motif and a corresponding domain.
Additional analysis can be also initiated from this webpage such as sequence alignment and comparative modeling of motif-domain complexes. This part is described in more detail at the end of this chapter as it may require some expertise in structural biology to be easily handled and truly fruitful. But most of the analyses using SLiMAn rely on the frontpage.
3.3.1 Parameter and filter description
We now briefly define the different filters available, while their specific use is illustrated in the next section.
Known PPIs can be selected from BioGRID or IntAct similarly to the precedent step (3.1.3). A heuristic scale (1-8) of confidence, combining several criteria for a SLiM-based PPI (predicted biophysical properties, experimental evidences...), was set to allow the user to quickly select PPIs with the highest level of confidence. Several types of filters can be applied :
- “ELM” and “PSP+” filters are defined respectivley to:
- Set the upper bound threshold for the ELM motif E-value, set ELM class of SLiMs (cleavage, modification, targeting, degradation, docking, ligand) or the ELM validated instances.
- Filter putative pairings based on the presence/absence of a given posttranslational modifications (PTM) and its requirement for the motif to be functional. As several SLiMs contain one or more PTMs, corresponding experimental information within each motif is made available through a link to the PhosphoSitePlus database (https://www.phosphosite.org/homeAction ).
- Values of structural parameters (IUpred2A, AlphaFold) or pair likeliness (HSM) can be set and tuned to refine displayed SLiMAn predictions of PPis.
- Text filters are applicable to limit the analysis to a given type of motif, domain or protein. To that end, the corresponding ELM or PFAM expression or the protein name can be input in the visual toolbox (e.g.: SH3_1 in the ELM box or SH3 in the PFam box).
3.3.2 Consulting validated instances of SLiM-based PPIs
The use of these different filters for interactomic data analyses is illustrated with a step-by-step and hierarchical approach which gradually defines the molecular features of the different PPIs found in a published interactome of the two human tankyrases (130 proteins) [30], members of Poly(ADP-Ribose) Polymerase proteins (PARPs) family, and their respective meta-interactomes from BioGRID-IntAct (170 and 80 interactants for TNKS1 and 2). The final results for those three searches in SliMAn can be found on the webserver (https://sliman2.cbs.cnrs.fr/study/TNKS1-2-Inter.html ; https://sliman2.cbs.cnrs.fr/study/TNKS1-Meta.html ; https://sliman2.cbs.cnrs.fr/study/TNKS2-Meta.html ). This approach aims at revealing potential SLIM-based interactions from the most likely to the less convincing ones while building a hierarchical molecular network.
Step 1: Identification of the most likely PPIs
- Request “Display all” within the Visualization panel.
- Select the ELM valid instances without applying any filter (i.e.: select “Validated Instances” within the ELM panel and switch off other parameters).
For the combined interactome TNKS1/2 [30], SLiMAn2 shows that 4 proteins would interact directly with tankyrase 1. These ELM valid instances involve a SLiM motif named Tankyrase Binding Motif (hereafter TBM) found in AXIN1, FNBP1, TERF1 and CASC3 and recognized by several ankyrin repeats (ARC or ANK) of TNKS1 and TNKS2 (Figure 5A) [37]. We can observe that these TBM motifs are predicted to be in unstructured regions (pLDDT < 60) and prone to fold upon binding (ANCHOR2 score > 0.4).
At this stage of analysis, no other type of SLiM-based PPIs appears to involve TNKS1 or TNKS2 while two other PPIs are revealed as ELM instances between GSK3b-AXIN1 and GSK3b-TP53. Here, two distinct linear motifs in AXIN1, its TBM (21-28) and its GSK3b docking site (383-389) would bridge GSK3b and TP53 to the tankyrases (see Note 4).
Note that within the BioGRID/IntAct meta-interactomes, 4 ELM instances (TBM detected in AXIN1, FNBP1, TB182 and TERF1) are found for TNKS1 and none for TNKS2. Furthermore, it shows only a partial overlap with the TNKS1/2 interactome with 3 common interactants (out of 5 in total).
Step 2: Detection of highly-confident SLiM-based PPIs
- Switch off the ELM instances and set the level of confidence to 8 with a low E-value (0.005). This increases the number of proteins displayed from 6 to 19 among 128 preys for TBNKS1/2. Fourteen proteins harbor ELM motifs putatively recognized by 7 PFam domains, which include 4 protein-kinase catalytic domains and 2 FHA domains on top of the Ankyrin repeats from the two tankyrases. Here, the ELM validated instance for CASC3 with TNKS1 is filtered out as no experimental evidence is recorded in BioGRID (requested Bio total > 0) or IntAct (requested IntAct+HuRI > 0) for that pair.
- Switch on the disordered parameters (Anchor > 0.4; Short and long Disorder > 0.4; pLDDT score < 60) to filter out a few motifs (15 out 202) and keep only most likely ones.
At this level of confidence and filtering, two additional substrates of human tankyrases appear (BABA1 and GO45) and they are connected to no other proteins. AXIN1 is still connected to tankyrases as well as to GSK3s and therefore TP53. The table highlights three other sub-networks, one corresponding to a multimer of the protein-kinase Chk2 (through its FHA motif and domain), one connecting MRE11 and nibrin (again through an FHA pair) and a last one connecting the protein-kinase STK26 to STRN4, STRP1, and CT2NL, due to various phosphorylation motifs and multiple experimental evidences from BioGRID and IntAct.
At a similar level of confidence and filtering, 10 PPIs involving 11 proteins are highlighted for the meta-interactome of TNKS1 whereas no additional PPIs was obtained for TNKS2. Interestingly, 5 proteins (AXIN1, FNBP1, GO45, TERF1 and BABA1) supposedly interacting with TNKS1 are common to the BioGRID/IntAct meta-interactome and the TNKS1/2 interactome.
As illustrated with this example, SLiMAn facilitates the identification of both direct and indirect connections or possible ternary complexes. At such a stringent filtering, interactions or pairings predicted by SLiMAn merely match already well-known interactions. However, lowering the stringency, may result in too many pairings for simultaneous inspection.
3. Use text-based filtering to focus on one given type of pairings:
- Input PFam query: TNKS
- Input ELM query: DOC_ANK_TNKS_1
to display only the pairings involving Tankyrases and the DOC_ANK_TNKS_1 motif
This selection leads to smaller table with one partner for TNKS2 and 6 for TNKS1 within the TNKS1/2 interactome. A similar trend is observed in the TNKS1 and TNKS2 meta-interactomes (with 6 interactants for TNKS1 and one for TNKS2).
4. Select
- Lower the confidence level to 6: three more TNSK2 partners (BCR, 3BP2 and TERF1) appear and only one (3BP2) for TNKS1 within the TNKS1/2 interactome, while up to 13 partners are found in the meta-interactome of TNKS1 and 8 for TNKS2. Among the 8 tankyrase binders within TNKS1/2 interactome, 6 are found also in the meta-interactomes of TNKS1 or TNKS2, and 3 of them (TERF1, 3BP2, BABA1) are shared by the three. This is still representing a tiny portion of all the preys listed in the various studies using human tankyrases as baits. This suggests that more pairings to the tankyrases may have to be characterized (or not) through SLiMAn interface by navigating at much lower stringency.
- Filter for predicted disordered using the above threshold remove only one validated partner (PAGE4) in TNKS1 meta-interactome [38], which can be brought back by increasing the AlphaFold pLDDT threshold to 65 (instead of 60). This pre-filtering analysis indicates the disorder parameters to select TBM from various Tankyrase partners.
Step 3: Using biophysical filters to predict additional binders at low levels of confidence.
As low confidence level can correspond to low disorder predictions and/or too few experimental evidences, one might want to counterbalance the low overall stringency by using parameters adapted to the particular pairings under scrutiny. As the TBM SLiM in ELM (DOC_TNKS_1) corresponds to highly flexible sequences, one can use rather stringent biophysical and structural features. Hence,
- Set IUPred2A and AlphaFold filters with high values (Anchor > 0.4; Short Disorder > 0.4; pLDDT score < 65).
These values were derived from those observed for the TBM detected at high confidence level (first 8 and then 6; see above). This should allow us to dig into the (meta-)interactomes in a discovery mode and to spot more tankyrase partners actually bound through a TBM.
- Decrease the level of confidence to 4 (from 6).
This reveals 21 putative interactors among the 128 preys (16%) of the TNKS1/2 interactome and 37 (out of 170, 22%) in the case of the TNKS1 meta-interactome and 24 proteins (out of 80, 30%) for TNKS2. Of note, 11 of those binders are potential new PPIs, whereas 10 are shared between TNKS1/2 and the two meta-interactomes and only 6 are found in the three interactomes.
- Decrease the level of confidence to 2 (from 4).
Elven additional potential partners show up for the TNKS1/2 interactome bringing the total number of potential partners to 32. The low confidence scores (2) for most of those additional pairs (8/11), come from the lack of supporting experimental data within BioGRID and/or IntAct database, as the thresholds for disorder are stringent (and yield a confidence score of 2 by themselves). 14 binders are found in TNKS1/2 and the meta-interactomes, whereas 18 are new PPIs, and 7 are common to the three interactomes.
It should be noted that a relatively small overlap is also observed between the two BioGRID/IntAct meta-interactomes with only 20 common proteins among the 62 potential TBM-dependant tankyrase binders.
At first, these additional partners are questionable, as most of these new preys were obtained by only one independent experiment. Accordingly, they could need additional validations to ensure they indeed correspond to direct binding to one of the two tankyrases. Here, SLiMAn allows to point out which protein within the whole interactome, and which region in these proteins to prioritize in order to confirm this pairing.
Step 4 : Adding alternative motif sequences in SLiMAn
The relatively small number of tankyrase preys detected as direct binders, so far, indicates that other interactions are possibly still missed even at low stringency. Such failures could be due to domain-domain interactions (that cannot be shown explicitly by SLiMAn), to indirect interactions (see above and below), to the presence of divergent TBM or direct interactions mediated by other type of motifs and associated domains. The latter case is very likely as TNKS1 harbors several proline-rich SH3 binding motifs. Beside FNBP1 which possesses a SH3 domain but is also harboring a TBM, 3 nexins found in the TNKS1/2 interactome, do possess functional SH3 domains, while we detected no other connections to tankyrases otherwise. TNKS1 also harbors a PP2B docking motif and an FHA recognition motif. However, the latter is not phosphorylated according to PSP+ and, therefore, may be considered as not functional (see below).
But one cannot exclude that the ELM motif is defined with a too stringent sequence signature. In fact, alternative motifs have been described in several substrates of tankyrases [39, 40]. Different from the stringent canonical TBM signature (DOC_ANK_TNKS_1: .R..[PGAV][DEIP]G.), the closely related (.R...[PGAV].G.) corresponds to a second motif with one additional residue within the same interacting partners [40].
Accordingly, search for potential alternative motifs that could fit into the TBM binding groove.
- Survey the crystal structures of tankyrase bound to various peptides
- Dig into the literature about tankyrase interactions.
Structural studies corroborated by directed mutagenesis and affinity measurements, point to the importance of an acidic residue in +2 position of the strictly conserved glycine [37]. These alternative TBMs may possess the new signatures R.{2,3}[PGAVSCT].G.[DE] or R.{3,4}[NDQEIVPT]G.[DE].
- Use the “Create your own RegEx” option to manually add a new signature to the initial query step in order to screen for additional Tankyrase substrates.
- Add the three patterns named respectively: Alt1 (.R...[PGAV].G.), Alt2 (R.{2,3}[PGAVSCT].G.[DE]) and Alt3 (R.{3,4}[NDQEIVPT]G.[DE]).
In the TNKS1/2 interactome, the addition of alternative patterns combined with the canonical ELM-Ankyrin signature increases the number of potential Tankyrase interactors from 32 to 46 (for Alt-1), to 41 (Alt-2) and 41 (Alt-3), respectively.
- Compare, in this particular case, the enrichment levels for ADP-ribosylated proteins to evaluate each signature (i.e.: presence of the protein in the ADPriboDB 2.0 database [41] ).
The proportion of ADP-ribosylated proteins increases from 48 % without filtering (complete interactome) to 65 % (ELM motif), 63% (Alt2) and 84% (Alt3) for each single filtering but for Alt1 (with only 43% of modified proteins). The best enrichment level (82 %) is obtained when combining the alternative sequence motifs Alt2 and Alt3 to the ELM canonical signature, which filter in 45 tankyrases substrates. Among these additional tankyrase partners, several were validated by low-throughput experiments (e.g.: 3BP2, 3BP5, RNF146). It also identified alternative TBM such as the second functional TBM in Pex14.
These results suggest considering alternative motifs for tankyrase recognition.
Step 5: Selection by ELM classes of SLIMs
To focus or hierarchize the search for other motifs, SLiMAn2 also offers the possibility to analyze PPIs for each ELM class type with variable E-values. This mode is quite convenient as it reduces the size of the table of ELM-PFam pairing. The rational for filtering by ELM class type is also based to the different intrinsic properties of the PPIs. Indeed, SLiMs leading to the most stable PPIs (e.g.: SH3) are mainly presented in “Docking” (DOC) and “Ligand”(LIG) class types whereas more transient SLiM-based PPIs are found in “Modification” (MOD), “Cleavage”(CLV) and “Targeting” (TRG). In addition, different SliMs have distinct tendencies for disorder and for folding upon binding.
1. Lower the confidence level from 8 to 2 to search for other likely direct PPis of Tankyrases in the TNKS1/2 interactome.
Similar disorder parameters than for the TBM-Ankyrin PPI were used but other filtering can be also set for each ELM-PFam pairs. For some DOC ELM classes (PDZ, SH3, SH2), SLiMAn integrates HSM biophysical prediction, enhancing filtering options [36].
2. Filter by name with “SH3” the two well-known interaction motifs of TNKS1.
In fact, the high-confidence interaction of FNBP1 with TNKS1 does not involve a TBM but a SH3 polyproline motif. Other SH3-based PPIs have lower levels of confidence (5) corresponding to three syntaxins (SNX9, 18 and 33).
3. Use HSM filters to rank the multiple pairing through SH3 motifs.
Precisely, 14 proteins are found to potentially recognize 13 SLiMs in TNKS1, from three different classes (LIG, DOC and MOD) and localized in three N-terminal highly disordered regions (1-10; 24-83 and 145-166). By filtering for LIG and DOC ELM class types, it remains 10 potential interactors with FHA (KIF1a, KIF1b, NBN, SLMAP, CHK2) and the already mentioned SH3 (FNBP1, SNX9, SNX18 and SNX33) as well as Metallophosphoesterase (MRE11) domains. From them, KIF1b, MRE11 are already directly connected via TBM motifs to the tankyrases. Of note, apart from FNBP1, none of these potential TNKS1/2 binders are present in TNKS1 or TNKS2 meta-interactomes. However, a favorable SH3 based PPI is also predicted in the TNKS1 meta-interactome between TNKS1 and UBS3B. For TNKS2, similar parameters reveal no direct SLiM-based PPI in the TNKS1/2 interactome as well as in the TNKS2 meta-interactome in agreement with the lack of disordered N-terminal part compared to TNKS1.
After similar step-by-step selections for the other SLiM class types (TRG, DEG, CLV), 17 new PPIs, composed of 4 direct SH3 PPIs with TNKS1 and 13 indirect (1 LIG, 2 DEG, 10 TRG) complete the TNKS1/2 interactome. Overall, 10 new direct or indirect SLIM-based partners have been added to the network on top of 42 proteins.
Step 6: PTMs and recognition of MOD class SLiMs
SLiMs and PTMs are tightly interconnected, although some protein modifications may occur due to chemical reactants with little site specificity (but for the modified residue) such as sulphur oxidation. By essence, most PTM sites should be associated with a SLiM, although not all have been precisely defined already [19]. Only a subset has been written in the "Modification" (MOD) class in the ELM database. These SLiMs are recognized by enzymes most likely through a transient interaction leading to the modification of one residue. Because of the transient nature of these interactions, we may not expect to detect them with most techniques dedicated to interactomics studies. Nevertheless, these modifications are often of uttermost importance for the functioning of macromolecules and need to be identified. Therefore, other validation schemes are required.
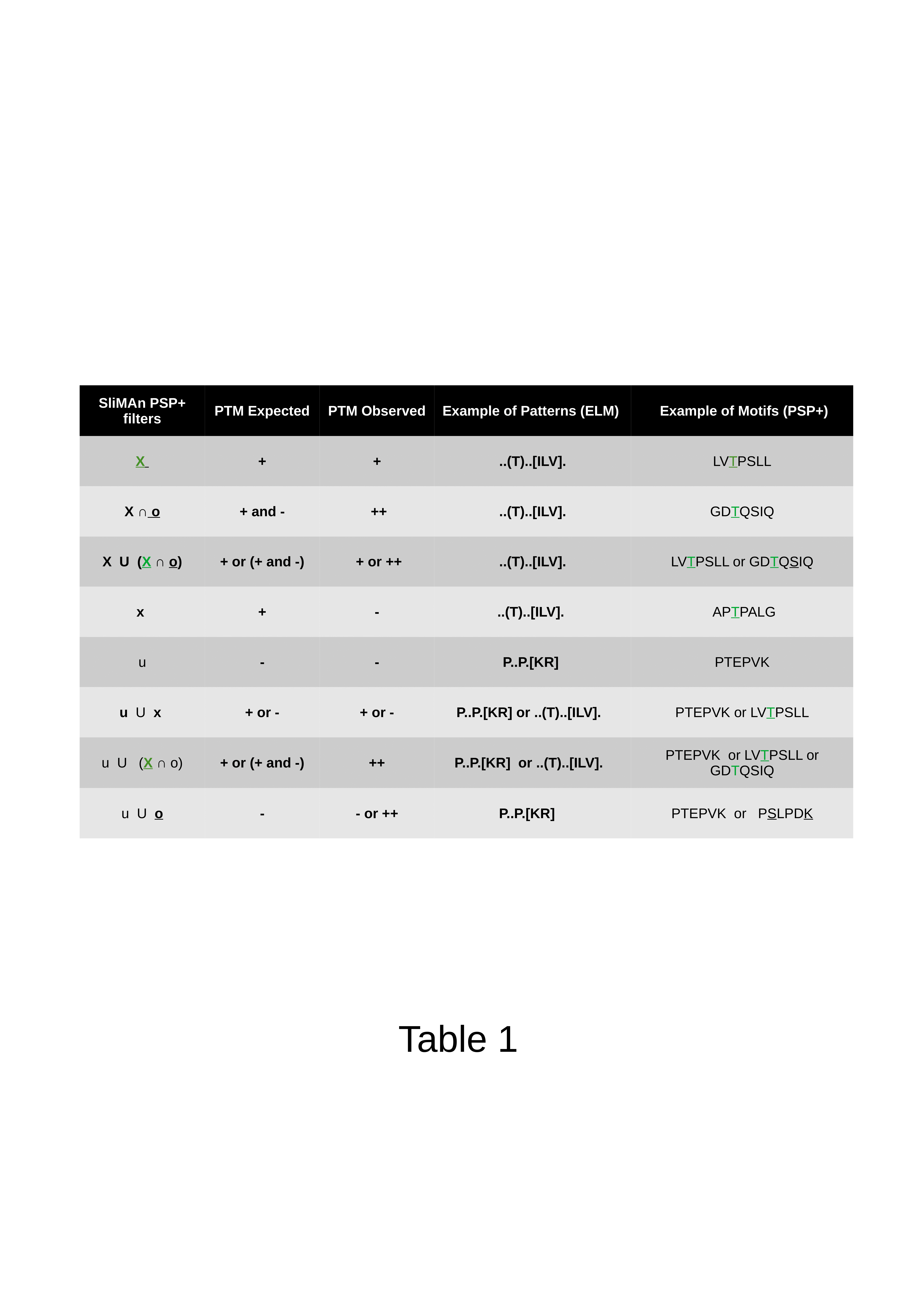
SLiMAn highlights the residue that should be modified for a given MOD motif. It also highlights any residue if a PTM has been annotated in the PSP+ database (for a small set of model organisms including mainly human and two rodents). A color code and a filtering scheme were set in the new version of SLiMAn (PTM observed or not) to ease the selection of the most favorable MOD SLiMs (Table 1).
Unfortunately, while ELM precisely defines the enzyme involved in those modifications (e.g.: MOD_CK2_1), the associated PFam domain comprises a large set of related proteins (e.g.: PF00069 and PF07714 for the majority of protein-kinases). Accordingly, SLiMAn is misled and frequently connects a motif with various enzymes for the same functional class ignoring the actual specificity, as illustrated below. This pairing should be cautiously considered when listed in a SLiMAn output.
- Filter for MOD by setting disorder: Anchor > 0.4; Short and long Disorder > 0.4; pLDDT score < 65), 16085 PPIs at confidence level 2 are highlighted in the TNSK1/2 interactome. Most E-values are weak (>0,01) and correspond to a high frequency motif sequences.
- Filter with PSP+ to select motifs for which the critical PTM has been experimentally detected (Table 1). The number of predicted pairings is 5735 among which 244 are supported by experimentally observed PPIs in BioGRID and/or IntAct database. These 244 PPIs involve 5 protein-kinases (TAOK2, CHK2, STK26 and GSK3Aa and GSK3B)b and 21 substrates (containing multiple motifs). In comparison, for the TNKS1 meta-interactome, similar filtering leads to 746 PPIs at confidence level 2 supported by 6 enzymes (STK11, STK36, TINIK, TITIN, PTEN and M4P4) and 28 substrates. For the TNKS2 meta-interactome, 3 kinases (PTEN, STK11 and MK01) and 8 substrates are potentially involved in 102 PPis. Whereas AXIN1, a well-known substrate of tankyrases, is present in the three interactomes, GSK3 kinases are however surprisingly absent, that is probably due to the higher prevalence of direct PPIs in the meta-interactomes, at least for the tankyrases. This observation is also supported by a very low number of indirect PPIs (only 2 for TNKS1) that have been found for the two tankyrases meta-interactomes.
- Use PSP+ information for additional filtering. Each motif should be scrutinized by navigating between SLiMAn and PSP+ database, which is easily accessible for each annotated motif PTM. Using PSP+ indications, two GSK3 phosphorylation sites on MCL1, already link to the tankyrases, can be validated. Indeed, a transient protein complex might bring together MCL1, tankyrases, AXIN1 and GSK3 kinases. Similarly, TP53 phosphorylation by CHK2 (T18) appear also highly likely.
Another illustration for the usefulness of these tools is the highly sophisticated scenario that links GSK3b to AXIN1, that can be anticipated with SLiMAn2. The prior link of GSK3b involved a previously selected docking SLIM-based PPI =. Furthermore, AXIN1 phosphorylation by GSK3b is itself phospho-dependent as the motif must be primed. Despite its low e-value (0,026), the corresponding motif can be validated as information from PSP+ confirms that the motif is phosphorylated at the two required positions (S75 and T79).
Overall, 10 MOD PPIs, involving 4 kinases and 5 substrates, have been selected in the TNKS1/2 interactome.
Step 7: Using PSP+ to filter other PTM-dependant PPis.
Several proteins (Kif1a, Kif1b, CHK2, NBN and SLMAP) in the TNKS1/2 interactome contain an FHA domain which recognizes a phosphorylated threonine in the LIG_FHA motif. As the e-value of the ELM FHA motif is quite high (>0,005), SLiMAn predicts a high number (3505) of putative FHA_1 based PPIs that can be further select :
- Switch off all ELM classes but the LIG one.
- Select motifs harboring a modified residue with PSP+ (Table1).
The number of SLiMs drops to 265 comprising 155 mono-modified and 110 multi-modified motifs.
- Apply BioGRID/IntAct databases filters
The number of potential FHA_1 motifs involved in known PPIs decreases to 24 (14 mono and 10 multi-modified motifs).
- Use PSP+ information for additional filtering
After a survey of these particular PTM-modified SLIMs, a total of 18 proteins, not interacting by a TBM, can be linked to tankyrases for the TNKS1/2 interactome. Interestingly, 61 % (11/18) of these additional partners are ADP-ribosylated suggesting that they do belong to the TNKS1/2 interactome.
Step 8: PTMs modulating SLiMs-based PPIs
Beside the MOD class (see step 6), all the other ELM classes may contain motifs involving modified residues. The modification can be mandatory for the recognition (designated as primary/mandatory) such as the phosphorylation of a tyrosine for a SH2 motif or a threonine for a FHA motif (see step 7). Alternatively, it may not be required (designated as secondary/accessory), although it may still interfere with any binding event. Some secondary modifications have been shown to be important functional switches, as mandatory ones are, but secondary PTM can be neutral, favorable or unfavorable to binding [42].
Accordingly, it is important to discriminate these two types of modifications depending on the motif under scrutiny. Hence, SLiMAn indicates, like for the MOD SLiMs, the PTM required for a given ELM motifs but also those detected in the PSP+ database. The color code and a filtering scheme differentiates the various situations (PTM observed or not; required or not), in support of more accurate searching for important motifs requiring PTMs or harboring secondary switches.
- Switch off all ELM classes but the LIG one.
- Select “no or accessory PTM” (u U o).
- Adjust disorder and confidence thresholds if necessary. Here, strict disorder is set on so that confidence can be very low (1).
This selection highlights the presence of a phosphorylation site (S432) in the TRF1 binding motif (LIG_TRFH1) of NBN, although this modification is not required. Experimental data listed in PSP+ indicate that this modification is rather frequent and deleterious for the interaction between NBN and TRF2, a paralogue of TRF1. This illustrates another example of the utility of PSP+ selection tools for PTM analysis.
3.3.3 Interactome network viewing using Cytoscape
Once a selection is achieved, it can be visualized in a dedicated Cytoscape window displaying the corresponding network. The progression of the analysis is illustrated in Figure 7.
By default, all the proteins harboring a validated pairing (ex.: TNKS-FNBP1) are connected. Each protein is shown as a purple rectangle that contains a green hexagon representing a PFam domain. A set of different colors characterizes each type of link between two protein partners: ELM motif/PFam domain pair, BioGRID or IntAct connections as well as HSM scoring. This potentially emphasizes dense sub-networks that usually correspond to macromolecular assemblies (based often also on domain-domain interactions) and/or singletons (not shown here) that may require further inspection. The latter may belong to the studied interactome - through unknown interaction - or correspond to spurious preys. Proteins can be rearranged within this window to better show these networking features. This may provide clues to resume searching for ELM/PFam pairing by focusing on particular proteins. This analysis is complementary to the analysis summarised in the main table.
For example, in Figure 7 several protein complexes, like STRIPAK (striatin-interacting phosphatase and kinase), MRN (MRE11, RAD50, NBN) that were initially disconnected or only lightly connected to the Tankyrases are linked (red arrows) at the end of the analysis. The addition of alternative TBMs appear to directly link to MRE11 as well as the MRN complex to tankyrase. Furthermore, apparently indirect binders like MCL1 and CHK2 are also directly linked to Tankyrases.
3.4 Structural model prediction of a given PPI
Finally, SLiMAn can check for the presence of related complexes in the PDB. It requires a folded domain matching the referenced PFam in the structure as well as a peptide (less than 35 residues) matching the desired ELM motif, in order to be considered a potential template for comparative modeling. For each class in ELM, an extraction from the PDB led to suitable templates for almost half of the possible ELM/PFam pairs, for a total of 5325 extracted templates.
SLiMAn gives first access to an interactive webpage (SLiM-ID) to handle paired sequence alignments for both the motif and the PFam domain. Then, comparative modeling can be submitted and the results can be visualized on a second webpage (SLiM-IM). Models can be downloaded for further study. They can also be tagged as validated or discarded to further assist the user in defining the interaction network in the main result page.
3.4.1. Sequence to structure alignments
If pre-extracted templates are available for comparative modeling of a given ELM-PFam pairing,
- access the sequence alignment interface, which is presented under the SLiM-ID environment (see Figure 8A).
- In SLiM-ID, first a summary of the paired ELM motif and the matched PFam domain sequences is highlighted on the top of the page. In addition, double alignments (of the motif and the domain sequences) with potential templates of complexes are performed using two different tools, MAFFT [43] and BLAST [44].
- ELM motif and PFam domain boundaries are directly extracted from their respective databases. If needed, manually edit them to re-compute alignments with re-defined sequence boundaries (Figure 8B). The ELM motif is generally well aligned to the corresponding peptide within the template thanks to the conserved ELM signature. The PFam domain may be aligned on much more divergent templates (< 35% of sequence identity). This might indicate that the overall fold is conserved but this might not include the binding site. In case of too low sequence similarity, cautiously discard the match. In this situation, use alternative approaches to predict the desired complex (e.g.: using HADDOCK [45] pepATTRACK[46] or AlphaFold [28]). Because SLiMAn requires a perfect match between the ELM motif regular expression to detect the peptide in a template, it may sometimes miss suitable templates. Again, alternative routes to modeling are necessary in that case.
- To guide the selection of the most suitable templates, several alignment metrics are computed: sequence identity (%ident), query coverage (%QueryCoverage), template coverage (%TemplateCoverage) and a conserved contact score (CCS and %CCS). At any time, the alignment table can be sorted according to one of these metrics (see Note 6). In addition, to facilitate the visual inspection of the alignments, residues belonging to the peptide-protein interface are coloured (green, orange and red) according to the contact distances (of 4.0, 5.5 and 7.0 Angstroms respectively).
- Before launching the modelling process, select the desired entries to serve as templates for comparative modelling, according to two options:
- a custom selection by checking the box on the left of each alignment in the table,
- by using the automated selection tools, to select top 5, non-redundant PDB, or all available templates.
Once, the alignments have been optimized, validated and at least one template is selected (Figure 8), click the “Launch modelling” button to start the comparative modelling process using SCWRL3.0 [47].
3.4.2. Structure Modeling
During the modeling process (approximately a few seconds per model) of the complex by SCWRL3.0, identical side-chains are kept fixed during the optimization first of the domain (in presence of the peptide from the original template), then of the peptide (in presence of the modeled domain).
- The completion of modelling, triggers a re-direction to the SLiM-IM environment. In the example, the 3Dmol.js viewer is used to display the complexes (Figure 9) [48]. In addition, an interaction analysis is performed by BINANA [49], highlighting favourable hydrophobic contacts (grey spheres) and hydrogen bonds (black arrows) as well as potential steric clashes (red spheres).
- At the bottom of the page is displayed a table containing the various information and intermediate models generated along the process. This table holds the original PDBid, its extracted SLiM-domain templates, model of the domain, model of the motif and the reconstituted complex. Click on the displayed structures to visualized or downloaded for local analysis.
- “Validate” or “Discard” models in the last column of the Table, based on own expertise.
- Click on the “Save selection” button, to erase discarded models and include validated models in the hit prediction table in SLiM-IP. The latter will be easily searchable using the “SLiMIM valid models” filter.
{kind=link}