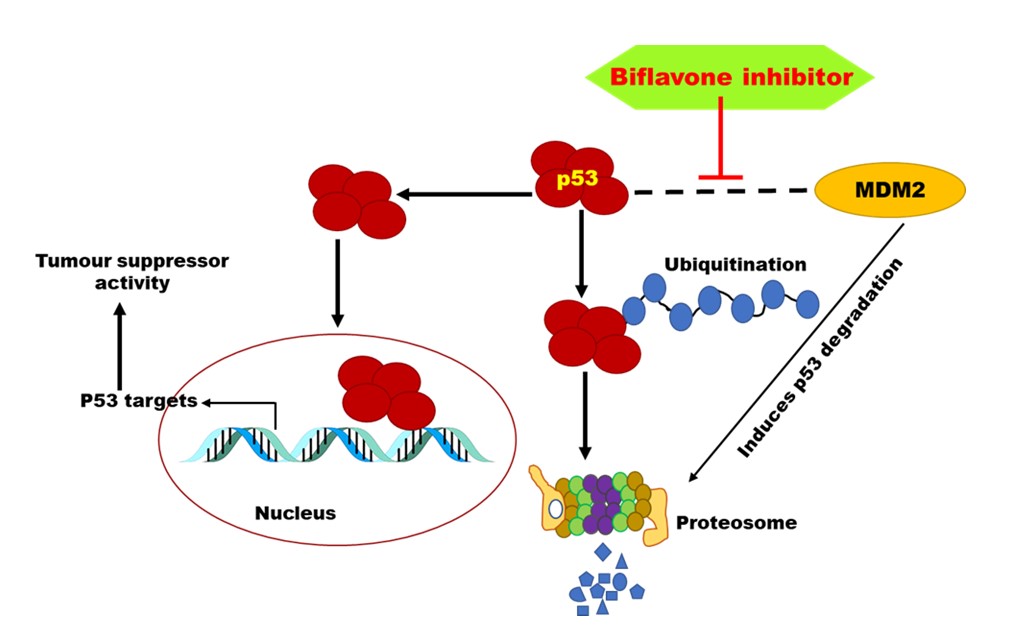
The MDM2 protein is a negative regulator of p53 and inhibit its transcriptional activity after binding, which in turn favouring unrestrained cell proliferation. Molecular docking studies of selected six bioflavonoids of C-C [amentofavone (1), robustafavone (2), agasthisfavone (3)] and C-O-C linkages [ochnaflavone (4), hinokiflavone (5), and delicaflavone (6)] along with a known inhibitor, nutlin-3 (7) against p53-MDM2 interaction were done to understand the underlying inhibitory mechanism of these biflavonoids.
Phe19, Trp23, and Leu26 amino acid residues at the hydrophobic pockets forming p53 “hot spot triad”, which are known to be participated in the binding interactions with the N-terminal domain of MDM2, which is referred as the p53-binding domain of MDM2 (Fig. S2 in the Supporting information, page S2) [25]. All of the C-C and C-O-C types biflavonoids effectively blocked the p53-MDM2 interactions due to its greater affinity for the p53 binding domain of MDM2, which eventually retarding MDM2 from interacting with p53, and thus, the p53-MDM2 interactions are disrupted. Same type of event is observed in case of the reference compound nutlin-3 (7), which is widely reported for its effectiveness to inhibit the interaction of MDM2 and p53 PPIs by blocking the p53-binding domain of MDM2 in the p53-MDM2 co-crystal (PDB ID: 1YCR) [33] as manifested from the MDM2-nutlin-3 docked structure (Fig. S1 in the Supporting information, page S2). Affinity and the strength of a specific biflavonoid ligand, which binds to the pocket of a target protein, can be expressed in terms of binding energy (ΔG) and the ligand having lower binding energy is preferred as a potential inhibitor.
3.1 Interaction of C-C type biflavonoids for MDM2 at its p53 binding domain
Docked structures MDM2 and C-C type biflavonoids like, amentofavone (1), robustafavone (2), agasthisfavone (3), are shown in Fig. 2(A), Fig. 3(A) and Fig. 4(A) respectively. All of the C-C type biflavonoids were found to bind at the site of MDM2, which was previously occupied by the p53 partially in the MDM2-p53 co-crystal (PDB ID: 1YCR). As shown in Table 1, the binding energy (ΔG) of nutlin-3 for the MDM2 active site was obtained as -7.4 kcal mol−1, while in the case of amentoflavone, the corresponding binding energy (ΔG) was found to be -8.7 kcal mol−1. These results reveal the strong affinity of amentoflavone (1) for MDM2 compared to nutlin-3. Interestingly, amino acid residues, GLN-72 and HIS-96 are found to participate in the interactions with amentoflavone through non-covalent bond formation, like hydrogen bonds (H-bonds) with the binding pockets of MDM2 protein. However, the value of binding energy of robustaflavone (2) and agathisflavone (3) with MDM2, were found to be slightly lower compared to the amentoflavone, although its higher compared to the reference compound nutlin-3. The binding energy (ΔG) of the robustaflavone and agathisflavone for MDM2 as calculated from their optimized docked structure was found to be -8.5 kcal mol−1 and -7.7 kcal mol−1 respectively (Table 1). It was also observed that no hydrogen bond (H-bond) formation between robustaflavone and agathisflavone with MDM2 shown in Fig. 3(A)-(i) and Fig. 4(A)-(i), and this can be the possible reason for its lower binding energy value. From the data of Table 2-4, it is also clear that amino acid residues and the bond distance involved in the docking between the MDM2-amentoflavone complex is different than those involved in the docking between MDM2-robustaflavone and MDM2-agathisflavone.
3.2 Interaction of C-O-C type biflavonoids for MDM2 at its p53 binding domain
MDM2 and C-O-C type biflavonoids like, ochnafavone (4), hinokifavone (5) and delicafavone (6) docked structures, are shown in Fig. 5(A), Fig. 6(A) and Fig. 7(A) respectively. All of the C-O-C type biflavonoids were found to bind at the same site of MDM2 like C-C type biflavonoids. As shown in the respective figures [Fig. 5(A), Fig. 6(A), Fig. 7(A)], the corresponding C-O-C type biflavonoids such as, ochnaflavone, hinokiflavone and delicaflavone, the corresponding binding energies (ΔG) were found to be -8.2, -8.2 and -7.2 kcal mol−1 respectively (Table 1). This also indicates the similar affinity of hinokiflavone and ochnaflavone for MDM2. However, the value of binding energy of hinokiflavone and ochnaflavone with MDM2, were found to be slightly lower compared to the amentoflavone, although its higher compared to the reference nutlin-3. The binding energy (ΔG) of the delicaflavone as calculated from their optimized docked structure was found to be -7.2 kcal mol−1, signifies the comparable affinity of delicaflavone and nutlin-3 towards the MDM2. It was also observed that the delicaflavone did not bind properly at the p53 binding pocket of MDM2 [Fig. 7(B)] and this can be the probable reason for its lower binding efficiency. From the data of Table 5-7, it is also clear that amino acid residues participated in the docking between the MDM2-hinokiflavone and MDM2-ochnaflavone complex are different than those involved in the docking between MDM2-delicaflavone.
3.3 Interaction of C-C or C-O-C type bioflavonoids towards free p53 and MDM2-bounded p53
Molecular docking studies elaborated so far established that both C-C and C-O-C type biflavonoids potentially bind with the MDM2 N-terminal domain but with different binding mode and efficiency. We also performed docking to show how MDM2 interacts with p53 when it is a part of the biflavonoid-MDM2 docked structure and how each biflavonoid interacts with p53 either in their free state or in the complex state with MDM2. We have explored the binding nature of MDM2 with the free p53 separated from MDM2-p53 co-crystal, which is bounded with either C-C or C-O-C type biflavonoid in the most stable docked structures. To validate the results, same PDB file of p53 was also subjected to the docking with free MDM2 protein. The part structure of separated p53 was considered as a rigid ligand by prohibiting the rotations of all the rotatable bonds to retain the structural compatibilities with the parent crystal structure after the docking. The binding energy (ΔG) value of the interaction between free p53 and the free MDM2 as calculated from their optimized docked structure was found to be -18.6 kcal mol−1 (Table S6 in the Supporting information, page S7), which is quite higher. During this docking, free p53 is docked at the same site, where it resided in the co-crystal of p53-MDM2 before its separation (Fig. S2 in the Supporting information, page S2) [26]. It also involved same amino acids which are participating in the hydrogen bonding interactions in the co-crystal of p53-MDM2. Interestingly, for both C-C and C-O-C categories of biflavonoids, the binding energy (ΔG) values were obtained from the docking between MDM2-biflavonoid complex and free p53 are lower compared to the free p53 and free MDM2 docking. Table S15-S20 in the Supporting information, page S10-S12 represents the corresponding docking scores. The binding energy (ΔG) values for all the biflavonoids of C-C and C-O-C categories lies between -10.3 to 11.5 kcal mol−1, which are very much lower compared to the binding energy obtained for the p53 and free MDM2 docking. This significant difference in the binding energy value is because of the fact that individual biflavonoid moiety was occupied at the p53 binding domain of the MDM2 which is considerably retarding the binding of p53 with MDM2. The p53 is retarded to get easy access to MDM2 to bind with it at its actual site due to the steric factor, which driven p53 to orient itself and bind at the different sites. Fig. 2(B), 3(B), 4(B), 5(B), 6(B) and 7(B) represents the possible bonding interactions between the separated structure of p53 and the MDM2-biflavonoid docked structures for both the C-C and C-O-C categories of biflavonoids. Amino acid residues involved in the docking between the free p53 and MDM2-biflavonoid complex for each C-C or C-O-C type biflavonoid are depicted in Table 8-13 respectively. Furthermore, from Fig. 7(A) and (B), it is observed that the delicaflavone (6) binds MDM2 somehow in a different position compared to the other biflavonoid. These may be the plausible reason behind the lower binding efficiency of delicaflavone in comparison to the other biflavonoids towards p53-MDM2 PPIs.
3.4 Drug-likeness prediction and pharmacokinetic properties evaluation
In drug discovery process, at the initial stage predicting the toxicological and pharmacokinetic profiling of any potential lead molecule is an effective method. Here, we investigated the absorption, distribution, metabolism, excretion and toxicological profile [ADMET] profiles of the C-C and C-O-C type biflavonoids and compared with the reference nutlin-3 compound using SwissADME [http://www.swissadme.ch/] [31] and admetSAR web servers [http://lmmd.ecust.edu.cn/admetsar1/predict/] [32]. Predicted results are presented in Tables S2-S3 in the Supporting information, page S5-S6. All the biflavonoids and nutlin-3 satisfied and fulfill the Lipinski’s rule of five [Table S2 in the Supporting information, page S5] as well as exhibited the most desirable pharmacological properties [31-32]. The result of the predicted pharmacokinetic properties of these biflavonoids are reported [Table S3 in the Supporting information, page S6], had satisfactory toxicity status due to their non-carcinogenic and non-mutagenic properties as well as their inability to inhibit the hERG gene encoding the potassium (K+) ion channels necessary for the normal electrical activity in the heart. It is also further supported by the fact is that, both the C-C and C-O-C type biflavonoids exhibited acceptable LD50 values [Table S3 in the Supporting information, page S6] and they were found to be in class II on the basis of their oral toxicity which is quite desirable. As revealed, biflavonoids and nutlin-3 [Table S3 in the Supporting information, page S6], are permeable to the human intestinal membrane and Blood Brain Barrier. Importantly, biflavonoids and nutlin-3 lacks the Caco-2 cell permeability. Both the compounds also showed good aqueous solubility. Moreover, compared to nutlin-3, all biflavonoids as lead compound, are non-inhibitor of the renal organic cationic transporter 2 which signifies the total clearance of the body system. From our observations, we can infer that both the C-C and C-O-C biflavonoids may act as lead compound and could be safe to prescribe as treatment option against cancer.
3.5 Molecular dynamics (MD) simulations
In order to analyse the behavioural mechanism i.e., stability, conformational changes and underlying molecular interactions of the candidate biflavonoid inhibitors on protein, the docked complexes of biflavonoids and nutlin-3 (reference compound) bound MDM2 protein were studied in a MD simulation in an explicit water solution for 40 ns using the GROMACS package (Ver 2022.4). Here, in our current study we choose amentoflavone (1) among C-C type and hinokiflavone (4) among C-O-C type biflavonoids as they are giving the higher docking scores. The trajectory plots representing the protein-ligand root mean square deviation (RMSD), the protein root mean square fluctuation (RMSF), the protein radius of gyration (RoG) and the solvent accessible surface area (SASA), are illustrated in Figs. 8-11.
3.5.1 RMSD analysis
RMSD plot of MDM2-Amentoflavone and MDM2-Hinokiflavone complex [Fig. 8 (a-b)], it is clear that from the beginning of the simulation process, both the complexes show higher fluctuation compared to free MDM2 protein and the respective biflavonoid itself. Despite of the deviation, the average RMSD value of the MDM2-Amentoflavone and MDM2-Hinokiflavone complex indicates the stability of the complexes. Although the RMSD value of MDM2-Amentoflavone is higher [2.5±0.5 Å] compared to MDM2-Hinokiflavone complex which remains within 1.5±0.5 Å. On the other hand, from Fig. S4 (a) in the Supporting information, page S3, it is visualized that despite the initial higher fluctuation of the protein-ligand complex, both free MDM2 and MDM2-nutlin-3 complex attains a stable state after 15 ns of MD simulation process and it remains stable till the end of the simulation.
3.5.2 RMSF analysis
One can easily predict the rigid or flexible nature of a particular complex from the Root Mean Square Fluctuation (RMSF) analysis. The RMSF plots of MDM2-Amentoflavone and MDM2-Hinokiflavone complex depicted in Fig. 9 (a-b), shows the initial fluctuations of the protein-ligand complex and thereafter, it remains at 1.5±0.5 Å for a long period of time, which shows the stability of the protein-ligand complex throughout. The RMSF values for free MDM2 and the corresponding biflavonoids remains less than 0.5 Å throughout the simulation. From Fig. S4 (b) in the Supporting information, page S3, it is clear that the RMSF value of the MDM2-Nutlin-3 complex, the RMSF value is about 0.50±0.25Å.
3.5.3 RoG analysis
The compactness of a system is generally measured with the help of radius of gyration (RoG). From the RoG plot of free MDM2 and MDM2-Nutlin-3 complex [Fig. S4 (c) in the Supporting information, page S3], it is clear that like previous observations (i.e., RMSD and RMSF), despite of initial fluctuations after 10 ns of molecular simulation, both free protein and protein-ligand complex indicates the stability of the protein-ligand complex. After 10 ns of simulation, the RoG of the protein-ligand complex (MDM2-Nutlin-3) shows a minimal fluctuation <1.5 Å. It again indicates the stability of the protein-ligand complex, like previous observations. On the other hand, both the MDM2-Amentoflavone and MDM2-Hinokiflavone complexes show some hump peaks, still, the deviation of the complex remains within 3.5±0.5 Å, which indicates its stable nature as a protein-ligand complex [Fig. 10 (a-b)].
3.5.4 SASA analysis
From the Solvent Accessible Surface Area (SASA) analysis, one can easily measure the surface area of a free protein/protein-ligand complex within a solvent system. SASA plot depicted in Fig. 11 (a-b), it is clear that from the beginning of the simulation process, both the MDM2-Amentoflavone and MDM2-Hinokiflavone complexes shows higher fluctuation compared to free biflavonoid, however, it remains stable till the 40 ns simulation process, which indicates the stability of the protein-ligand complex. Similar phenomenon is observed for the free MDM2 and MDM2-Nutlin-3 complex [Fig. S4 (d) in the Supporting information, page S3]. This is in agreement with the previous observations (RMSD, RMSF, and ROG).
{kind=link}