Quinabactin (QB, 3) is an important ABA functional analogue without ABA-like structure and has great application potential in agriculture. In order to address the key factors affecting the binding mode of quinabactin to ABA receptors and plant phenotypes, several QB analogues with fine-tuned lactam ring and sulfonamide group were designed and synthesized. Their effects on plant phenotypes, such as seed germination, seedling growth, stomatal movement and drought tolerance, were screened. Meanwhile, their binding affinity to ABA receptors, inhibitory activities on HAB1 phosphatase and ABA-response gene regulation abilities were evaluated. The results showed that their affinities to ABA receptors displayed subtle differences and were highly consistent with their influence on plant phenotypes. DHQB increased the inhibitory activity of QB on rice leaf growth by nearly one fold, showed that the weak activity of QB on monocots could be improved by its structure optimization. The change of sulfonamide orientation had the greatest influence on receptor binding and apparent bioactivities, whereas the poor activity of TFTQB (25) reflected the binding boundary between these analogues and ABA receptors. The change of a single factor on lactam ring had little effect on all activities, although activities were all slightly decreased. These effects were obviously due to the differences in their binding to ABA receptors, which had been reasonably explained by the molecular docking model. This study provided constructive insights for the development of novel quinabactin analogues.
Research Article
Structural fine-tuning analogues of quinabactin revealed the differences of their binding microenvironment with ABA receptors and their influence on plant phenotypes
https://doi.org/10.21203/rs.3.rs-3995941/v1
This work is licensed under a CC BY 4.0 License
You are reading this latest preprint version
Abscisic acid
Quinabactin ABA functional analogue
ABA receptor
Molecular docking
Abscisic acid (S-(+)-ABA, 1) is a chiral sesquiterpene compound, which was discovered in the 1960s by activity-oriented identification of plant growth regulators (Ohkuma et al., 1963). In addition to its role in defending cell physiology, ABA also mediates other abiotic stress responses (such as frost resistance), and plays a central role in inducing seed dormancy, controlling root structure and influencing several biological interactions (Cutler et al., 2010; McCarty, 1995; Yamaguchi-Shinozaki and Shinozaki, 2006; Zeevaart, 1998). Therefore, it has great potential in agricultural applications, for example, in berry coloring (Ban et al., 2000) and helping crops resist abiotic stresses such as drought (Teng et al., 2014), cold (Valluru et al., 2012) and salt (Munns et al., 2006). However, due to the fragility of its structure (inactivated owing to easily isomerizing in vitro (Plancher, 1979) and metabolizing in vivo (Oritani and Kiyota, 2003)) and difficult to synthesize, it has always been the goal of scientists for structural modification to obtain more powerful ABA functional analogues. Unfortunately, most of early researches in the field are not targeted accurately because ABA receptor has been unknown for nearly a half-century. Until 2009, the ABA receptors were identified by chemical genetics (Park et al., 2009) and yeast two-hybrid screening (Ma et al., 2009), and the early signal transduction mechanism basing on the binding of ABA receptor was clarified, making it possible for receptor structure-based rationally ligand designing. With the discovery of non-ABA-mimicking ligands, such as pyrabactin and quinabactin, and combined with ABA analogues discovered earlier, we can gradually outline the interaction modes between different ligands and various ABA receptors. However, this is far from enough.
ABA functional analogues can be divided into ABA analogues and non-ABA analogues. The former is a class of compounds obtained through structural optimization based on ABA structure, and they have almost the same action mechanism as ABA, such as 8′,8′,8′-trifluoro-ABA (Kim et al., 1995), 8′-methylidyne-ABA (Benson et al., 2015), PhABA (Nyangulu et al., 2006), iso-PhABA (Han et al., 2017), and so on. However, these compounds are relatively structural complicated, and the photoisomerization of conjugated side chain has not yet been addressed. Non-ABA analogues are completely separated from the ABA scaffold, which differs from the former in that they can only bind to some ABA receptors as agonists or even antagonists.
Pyrabactin (2) is the first non-ABA functional analogue screened by chemical genetics, which leads to the identification of ABA receptors in Arabidopsis thaliana and the elucidation of early signal transduction mechanism of ABA (Park et al., 2009). Pyrabactin is a selective agonist of PYR1 and PYL1, and a weak antagonist of PYL2 which activates ABA-like response in seeds, but has little response in vegetative tissues (Okamoto et al., 2013; Cao et al., 2013). In 2013, Okamoto et al. (Okamoto et al., 2013) and Cao et al. (Cao et al., 2013) screened out a sulfonamide compound quinabactin (3, QB, also known as AM1) independently with high ABA activity from the compound database almost simultaneously by different methods, which can activate some ABA receptors insensitive to pyrabactin and selectively bind to receptors such as PYR1, PYL1-3, PYL5 and PYL7 of A. thaliana. Its binding mode is very similar to that of ABA, showing that these receptors play a leading role in regulating ABA activity compared with other monomer receptors. Biological evaluation showed that QB (AM1) showed similar effects to ABA in all living biological tests, including inhibiting seed germination, promoting stomatal closure, reducing leaf water loss and improving drought tolerance of plants. Gene expression assay showed that QB could induce the expression of almost all ABA responsive genes. The crystal structure of QB(AM1)-PYL2-HAB1 complex shows that the carbonyl group of QB (AM1) plays the role of carbonyl group on ABA cyclohexenone ring and can form hydrogen bond with R120 and P92 on receptor and W385 on HAB1. Sulfonamide plays the role of ABA carboxyl, which can interact with some amino acids at the bottom of receptor binding pocket, thus anchoring QB (AM1) in the binding cavity.
The discovery of pyrabactin and quinabactin opened the door for the study of non-ABA functional analogues, and some QB (AM1) analogues or derivatives were used for preliminary structure-activity relationship evaluation. Cao et al. (Cao et al., 2017) introduced fluorine atoms and chlorine atoms into the 4-methyl benzene ring of QB (AM1) to provide derivatives 4–9, which can effectively inhibit the germination of Arabidopsis seeds and promote stomatal closure. Phosphatase experiments showed that these analogues could inhibit the activity of HAB1, and all of them were superior to QB (AM1). In the drought resistance experiments of A. thaliana and soybean, compound AMF4 (8, TFQB in this paper) showed the highest and most lasting activity, and its drought resistance effect was obviously better than ABA and QB (AM1). The crystal structure of PYL2-AMF4-HAB1 ternary complex shows that the hydrogen bond number formed by AMF4 and PYL2 is more than that of QB (AM1) and PYL2 due to the introduction of fluorine atoms, so that its affinity is obviously improved, thus possessing higher biological activity. Elzinga et al. (Elzinga et al., 2019) preliminarily clarified the limited substitutability of the substituent by substituting (10–18) the methyl group on the benzene ring. In order to obtain ABA functional analogues with simpler structure and better water solubility, Vaidya et al. (Vaidya et al., 2017) further simplified the structure of QB (AM1) by adopting the principle of electron isostere, replacing the carbonyl group in QB (AM1) with cyano group and replacing the original propyl group, thus obtaining the compound cyanabactin (19). Cyanabactin has the same activity as QB (AM1) in inhibiting seed germination, promoting stomatal closure and inducing ABA response gene expression. The discovery of cyanabactin proves that the bicyclic structure in QB (AM1) is not necessary to maintain the agonistic activity of ABA, which provides a new idea and inspiration for designing new ABA functional analogues with simpler structure and more valuable for agricultural production. Recently, Vaidya et al. (Vaidya et al., 2019) discovered the compound Opabactin (20) with excellent activity based on virtual screening, which is the first non-sulfonamide ABA functional analogue with great significance.
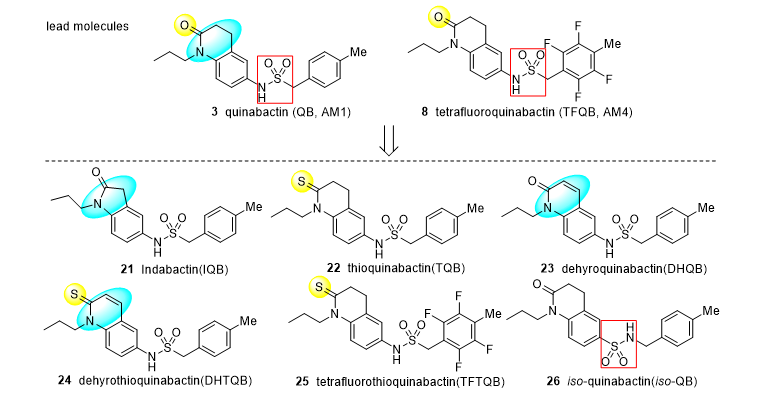
The discovery of these functional analogues of ABA with great structural differences shows that the ligand-binding pocket of ABA receptors has certain tolerance to structural changes of ligands. However, the related information is very limited at present, and the boundary of this tolerance has yet to be further revealed. In this study, we designed and prepared several QB (AM1) analogues (21–26). Collectively, as Scheme 1 showed, indabactin (IQB, 21) and dehydro-quinabactin (DHQB, 23) were designed to probe the importance of the size and coplanarity of lactam ring for QB (AM1, 3). Sulfur atoms, as isosteres of oxygen atoms, have similar chemical properties, but the atomic radius of sulfur atoms is larger than that of oxygen atoms, which can lead to changes in the interaction when binding to the receptors. Thioquinabactin (TQB, 22), dehydrothioquinabactin (DHTQB, 24) and tetrafluoroquinabactin (TFTQB, 25) were designed to evaluate the influence of the volume and hydrogen bond factors of lactam moiety. Furthermore, functional group interconversion is a common strategy in novel drug development (Jones, 2020). Based on this principle, iso-quinabactin (iso-QB, 26) was designed to reveal the linchpin of sulfonamide orientation. These subtle structural changes well revealed the structural requirements of ligand binding pockets for QB analogues, and had important guiding significance for the design of this kind of ABA functional analogues.
2.1. Chemistry
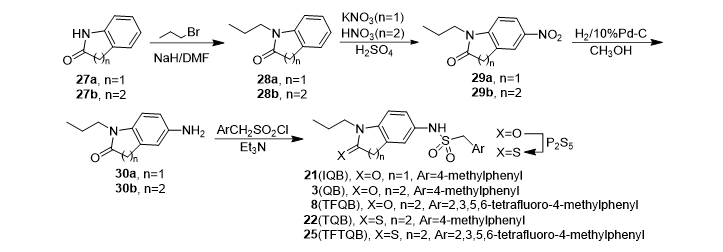
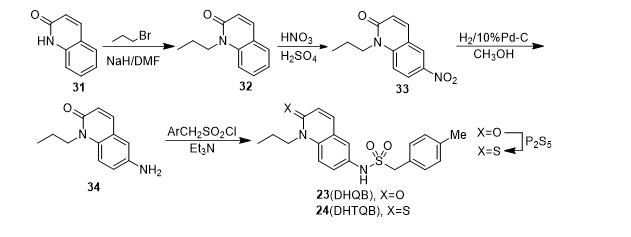
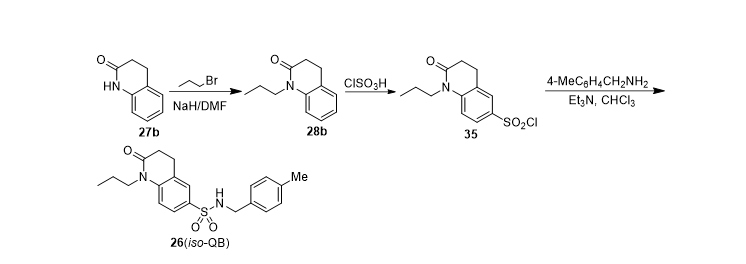
In addition to the different starting materials used, compound IQB, TQB, TFTQB, DHQB and DHTQB were conveniently synthesized by similar methods and the general pathways were illustrated as Scheme 2 and 3. Whereas iso-QB was prepared by chlorosulfonation of 32 followed by aminolysis (Scheme 4).
2.2. Bioassays
2.2.1 Seed germination inhibition activity
As shown in Table 1 and Fig. 1, all the synthesized compounds including positive controls exhibited good to excellent inhibitory activities on Arabidopsis and lettuce seed germination. At first, ABA exhibited the best inhibitory activity on both A. thaliana and lettuce, First of all, the inhibitory activity of ABA on A. thaliana and lettuce was the best. The IC50 for inhibiting seed germination of A. thaliana and lettuce could reach 0.20 µM and 1.34 µM. Secondly, DHQB (22), TQB (23) and DHTQB (24) showed the same level inhibition as QB (3) and TFQB (8) for both seeds, suggested that the single factor change of coplanarity (23) or volume (22) only caused a slight decrease of germination inhibiting. Thirdly, the activity of five-membered lactam (IQB, 21) was decreased by half compared to QB, which implied that six-membered ring was more suitable to inhibitory activity of QB analogues. Fourthly, the activity of TFQB (8) was higher than that of QB (3), indicating that the substitution of fluorine atom on benzene ring was beneficial to the activity, which was consistent with the results reported (Cao et al., 2017). Dramatically however, when TQB (23) was tetrafluorinated, the inhibition activity of TFTQB (25) was not only greatly reduced compared to TFQB (8), but also reduced by 2–3 times compared to TQB (23). Obviously, fluorination amplified the negative effect of sulfur atomic volume existing in TQB (23). At last, the activity of sulfonamide oppositely oriented isomer (iso-QB, 26) of QB was 5–6 times lower than that of QB, strongly demonstrating the central role of sulfonamide moiety in QB analogues.
| Compd. | IC50 (µM) | |
|---|---|---|
| A. thaliana | Lettuce | |
| ABA(1) | 0.20 ± 0.02 | 1.34 ± 0.11 |
| QB(3) | 1.33 ± 0.10 | 2.62 ± 0.19 |
| TFQB(8) | 1.15 ± 0.09 | 2.29 ± 0.18 |
| IQB(21) | 2.36 ± 0.13 | 7.13 ± 0.35 |
| TQB(22) | 1.53 ± 0.16 | 3.43 ± 0.28 |
| DHQB(23) | 1.39 ± 0.11 | 3.10 ± 0.25 |
| DHTQB(24) | 1.33 ± 0.15 | 2.98 ± 0.22 |
| TFTQB(25) | 4.98 ± 0.31 | 7.09 ± 0.57 |
| iso-QB(26) | 6.42 ± 0.41 | 14.56 ± 0.86 |
2.2.2. Seeding growth inhibiting activity
The inhibitory activity of synthesized compounds on the growth of A. thaliana and rice seedlings was tested and the results were summarized in Table 2. IQB (21), TQB (22), DHQB (23) had the same inhibitory effect on the root growth of dicotyledonous A. thaliana as QB, but the fresh weight inhibitory activity decreased. However, the inhibitory activity of five-membered lactam (IQB, 21) on A. thaliana fresh weight and rice leaf growth was poor. TQB (22) as thiolated analogues of QB (3) significantly decreased inhibition rates on rice leaf growth. This result strongly suggested that TQB could be used as a probe to study the seedling growth differences of monocots and dicots. However, DHQB increased the inhibitory activity of QB on rice leaf growth by nearly one fold, showed that the weak activity of QB on monocots could be improved by its structure optimization. Surprisingly, TFQB (8) exhibited enhanced inhibitory activity on rice leaf growth, but significantly decreased activity on root length and fresh weight of A. thaliana. The inhibitory activity of TFTQB (25) was deceased not only on A. thaliana, but also greatly on rice seedling growth. Obviously, fluorination amplified the negative effect of sulfur atomic existing in TQB (22). Not surprisingly, the inhibitory activity of iso-QB on seedling growth was also very poor.
| Compd. | A. thaliana (inhibition ratio %, 25 µM) | Rice (Leaf growth inhibition) | |
|---|---|---|---|
| Root length | Fresh weight | IC50 | |
| DMSO | 0 | 0 | / |
| ABA(1) | 66.27 | 63.04 | 1.50 ± 0 .19 |
| QB(3) | 72.26 | 65.20 | 5.98 ± 0.42 |
| TFQB(8) | 23.75 | 34.72 | 2.13 ± 0.18 |
| IQB(21) | 73.05 | 48.15 | 11.22 ± 0.63 |
| TQB(22) | 70.46 | 58.10 | 26.96 ± 0.75 |
| DHQB(23) | 73.85 | 56.48 | 3.29 ± 0.26 |
| DHTQB(24) | 62.48 | 65.51 | 5.91 ± 0.42 |
| TFTQB(25) | 2.40 | 16.36 | 30.29 ± 0.86 |
| iso-QB(26) | 41.92 | 26.77 | 9.89 ± 0.38 |
2.2.3. Plant drought tolerance induction
Wheat, soybean and A. thaliana were used as the representatives of monocots, dicots and model plants to measure the drought resistance induced by synthesized compounds. As Table 3 and Fig. 3 to 4 indicated, these compounds could induce drought resistance of plants in different degrees, and the wilting degrees of plants were in sharp contrast with DMSO group. In general, IQB (21), DHQB (22), TQB (23), DHTQB (24) exhibited similar drought-resistant induction abilities to ABA (1), QB (3) and TFQB (8), whereas TFTQB (25) was obviously worse than them. The water content analysis in soil showed that foliar spraying of these agents was helpful to maintain soil moisture, which was consistent with their drought resistance abilities (Table 3). Iso-QB (26) only showed weak drought-resistant induction activity, which showed that the reversion of sulfonamide orientation was also unfavorable to this performance.
| Compd. | wheat | soybean | A༎ thaliana | ||||
|---|---|---|---|---|---|---|---|
| gradea | water content in soil | gradea | water content in soil | gradea | water content in soil | ||
| DMSO | 0 | 18.31 ± 0.63 | 0 | 18.67 ± 0.89 | 0 | 11.32 ± 0.62 | |
| ABA(1) | ++++ | 37.62 ± 0.70 | ++++ | 32.77 ± 1.06 | ++++ | 25.61 ± 0.74 | |
| QB(3) | ++++ | 36.34 ± 1.13 | ++++ | 33.27 ± 1.01 | ++++ | 24.01 ± 0.65 | |
| TFQB(8) | ++++ | 36.47 ± 1.03 | ++++ | 33.92 ± 0.85 | ++++ | 27.14 ± 0.85 | |
| IQB(21) | ++++ | 36.01 ± 1.25 | ++++ | 32.47 ± 1.30 | ++++ | 24.67 ± 0.70 | |
| TQB(22) | +++ | 34.70 ± 0.95 | ++++ | 32.83 ± 0.99 | ++++ | 24.75 ± 0.71 | |
| DHQB(23) | +++ | 35.41 ± 0.97 | ++++ | 33.19 ± 0.87 | ++++ | 26.07 ± 0.69 | |
| DHTQB(24) | +++ | 35.15 ± 0.91 | ++++ | 31.53 ± 0.95 | +++ | 23.55 ± 0.85 | |
| TFTQB(25) | ++ | 27.65 ± 1.11 | ++ | 24.23 ± 1.11 | ++ | 20.70 ± 0.90 | |
| iso-QB(26) | + | 21.41 ± 1.05 | ++ | 24.87 ± 0.86 | + | 15.74 ± 0.54 | |
| a Drought resistance is graded by leaf wilting on the 7th day of drought treatment. “0”= no drought resistance activity; “+”= most of the leaves curled or wilted; “++” = more than 50% of the leaves curled or wilted; “+++”= a few leaves curled or wilted; “++++”= very few leaves curled or wilted. | |||||||
2.2.4. Induction of stomatal closure in A. thaliana
The transpiration can reduce the leaf surface temperature by evaporating and cooling, so reducing transpiration by inducing stomatal closure will lead to an increase in surface temperature. As shown in Fig. 5 and supplementary Table S1, after 24 hours of treatment, the stomata of DMSO group opened normally, and the leaf surface temperature was 21.49 ℃. In comparison, all QB analogues except iso-QB (26) displayed higher surface temperature not only than DMSO but also ABA treatment group (Fig. 5B). The results were consistent with their drought-resistant induction activities, showing that these compounds might have great potential in drought management. Among them, TFQB (8) treatment group reached the highest temperature, followed by QB, which was higher than other analogues by more than 0.3 ℃, indicating that these structural modifications to QB were unfavorable to reduce transpiration except TFQB (8), although they exhibited similar drought-resistant induction activities as above mentioned. This result also suggested that the improvement of drought resistance might not only depend on reducing transpiration. Interestingly, IQB (21) showed the comparable activity as TQB (22), DHQB (23), DHTQB (24) and TFTQB (25), which was quite different from its inhibitory activity, indicated the difference in recognition of lactam ring size between inhibition process and stomatal closure induction and IQB could be used as a good probe to study this difference. In addition, iso-QB (26) performed the worst again, indicating that reversed sulfonamide orientation was indeed unfavorable in recognition of ABA receptors.
2.2.5. Receptor binding affinity
The binding of ABA and its analogues to ABA receptor was the first step of early ABA signal transduction, and the binding strength directly affected their agonistic/antagonistic effects. However, there were many ABA receptors in plants (e.g. 13 receptors in A. thaliana), and their unique functions had not been accurately addressed so far. In order to verify the relationship between these receptors and the apparent biological activities, we measured the binding affinities of QB analogues to 10 ABA receptors of A. thaliana (except PYL7, PYL11 and PYL12) using microscale thermophoresis (MST). The Kd values and binding curves were summarized in Table 4 and supplementary Fig. 1. First of all, like QB and TFQB, these analogues were also selective agonists of group III and PYL5 receptors, indicated that they initiated ABA response signals in exactly the same way. For different receptors, the binding affinities with PYL5 were highly in line with the activities of stomatal closure and drought resistance induction. On the contrary, PYL3 might be the receptor with the least correlation with stomatal regulation, because all QB analogues show lower affinity than ABA.

2.2.6. Molecular docking analysis
To investigate the potency and specificity of QB analogues toward PYL2 and PYL5, we applied molecular docking to study the binding affinities and binding modes. On this basis, the optimized molecules were flexibly docked with the ABA receptors and the docking results were shown in Table 5.
| Compd. | Total-Score | Crash | Polar | CScore | Similarity | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PYL2 | PYL5 | PYL2 | PYL5 | PYL2 | PYL5 | PYL2 | PYL5 | PYL2 | PYL5 | |||||
| ABA (1) | 6.71 | 6.64 | -1.41 | -0.85 | 2.92 | 4.45 | 4 | 4 | 0.71 | 0.59 | ||||
| QB (3) | 5.30 | 5.61 | -4.88 | -1.26 | 0.05 | 0.87 | 3 | 1 | 0.62 | 0.56 | ||||
| TFQB (8) | 4.88 | 6.09 | -6.72 | -6.17 | 1.25 | 2.91 | 5 | 3 | 0.51 | 0.59 | ||||
| IQB (21) | 4.35 | 5.09 | -4.73 | -1.13 | 0.04 | 1.10 | 4 | 2 | 0.56 | 0.50 | ||||
| TQB (22) | 3.24 | 5.04 | -6.69 | -4.36 | 0.06 | 1.10 | 3 | 4 | 0.63 | 0.53 | ||||
| DHQB (23) | 3.36 | 5.35 | -5.47 | -5.55 | 0.56 | 2.56 | 5 | 4 | 0.53 | 0.58 | ||||
| DHTQB (24) | 3.50 | 5.61 | -6.65 | -1.55 | 0.05 | 0.22 | 5 | 2 | 0.53 | 0.54 | ||||
| TFTQB (25) | 2.49 | 4.05 | -9.61 | -2.58 | 0.07 | 0.22 | 3 | 2 | 0.58 | 0.54 | ||||
| iso-QB (26) | 3.60 | 3.80 | -7.02 | -3.25 | 0.01 | 0.00 | 3 | 3 | 0.51 | 0.50 | ||||
| Total Score is to evaluate the binding affinity between ligands and receptors. The unit of Total Score is -lgKd. Crash indicates the degree to which the ligand penetrates the protein, and Polar indicates the ability of an atom to form a hydrogen bond; Cscore is the comprehensive evaluation of five scoring functions in Sybyl, and the value represents the type of optimal scoring function;Similarity indicates the similarity between the docking compound and the original ligand ABA in the protein crystal structure. | ||||||||||||||
The docking results in Table 5 showed that the value of docking score was consistent with the activity of inhibitor, and the corresponding micro interaction mode (Fig. 7) was the decisive factor affecting its activity. We chose the following system for discussion. From the interaction mode between compounds QB (3) and iso-QB (26) and PYL2 (Fig. 7A), it could be seen that the sulfonamide group of QB (3) formed strong hydrogen bonds with protein residues Lys64 (2.6 Å) and Asn173 (2.3 Å), and the sulfonyl group also formed a strong hydrogen bond interaction with the surrounding water molecules. The sulfonamide group of iso-QB (26) only formed hydrogen bond interaction with residue Ser96 (1.8 Å). It was obvious that the strong hydrogen bond interaction was the reason for the strong inhibitory activity of QB to PYL2. For IQB (21), there was no interaction with the key residues of the active pocket in its action mode, and the sulfonyl group of IQB (21) was located at the inlet of the active pocket, so it did not show good inhibitory activity.
For QB (3), TFQB (8), DHQB (23) and DHTQB (24) with similar activity, it could be seen that they formed similar hydrophobic interaction with the receptor protein and existed in the active pocket in a conformation with similar folding degree. Although halogen atom F existed in the structure of TFQB (8), due to the lack of O, N and S atoms with Lewis basicity around the active pocket, there was no halogen bond interaction. Therefore, the inhibitory activities of the above molecules were also very similar.
From the interaction mode with PYL5 (Fig. 7B), we could also find that the phenyl of QB (3) tended to form van der Waals interaction with the hydrophobic residues Leu145, Val118 and Phe184 at the bottom of the active pocket. Due to the high rotatability of NH bond, the conformation of the phenyl connected to it was reversed by 90 degrees, so that the conformation of iso-QB (26) was folded to a large extent. At the same time, the hydrophobic environment of the phenyl was also changed, mainly forming hydrophobic interaction with Val185 and Ile188. Due to the change of conformation, the hydrophobic interaction was weakened, resulting in the decrease of activity. From the interaction modes of QB (3), IQB (21) and PYL5 proteins, it could be found that the carbonyl oxygen on the six membered ring of QB (3) could form three strong hydrogen bonds with residues Arg107 and Phe136, while IQB (21) could only form two hydrogen bonds with residues Lys87 and Arg107, and there were more hydrophobic amino acids near the six membered ring of QB, which could form a strong van der Waals interaction. DHQB (23), DHTQB (24) and QB (3) had similar bioactivity to PYL5, and their microscopic interaction modes also showed that the molecules had similar conformations in the protein active pocket, and the carbonyl O of DHQB (23), QB (3) and S of DHTQB (24) could form similar hydrogen bonds with residues Lys87 and Arg107.
2.2.7. Inhibition of HAB1 phosphatase activity
After binding with the receptor, ABA receptor agonist would form a binding interface with PP2C phosphatase (such as HAB1) and inhibit its activity, thus releasing SnRK2 and generating ABA response. However, an ABA receptor antagonist couldn’t initiate downstream signals after binding with the receptor. Therefore, the determination of phosphatase activity could be used to judge whether these compounds were ABA receptor agonists or antagonists, and this was done for all above mentioned receptors. ABA binding to all receptors can well inhibit the phosphatase activity of HAB1, so ABA is a broad-spectrum ABA receptor agonist. The complexes of QB, TFQB, IQB, TQB, DHQB and DHTQB with PYR1, PYL1-3 and PYL5 all showed strong inhibition to HAB1, while the complexes with other receptors poor, completely consistent with the above receptor affinity results, which suggested that they were strong agonists of these receptors. In addition, this also suggests that PYR1, PYL1-3, and PYL5 may be the key receptors for controlling seed germination, seedling growth, plant drought resistance and stomatal movement. In the above binding affinity experiment and inhibition of HAB1 phosphatase activity, the decrease of affinity of TFTQB and iso-QB to the receptors directly lead to the decrease of PP2C phosphatase inhibition activity.
2.2.8. Gene expression induced by QB analogues
The MST and phosphatase activity inhibition results had proved that these QB analogues could activate the core link of ABA signal pathway. To further verify the integrity of them in activating ABA signal pathway, we measured their effects on the expression of ABA response gene, including MAPKKK18, RD29A, RD29B and ABF3, which were ABA stress-related genes and could be induced to express by ABA (Yoshida et al., 2021). The results of RT-PCR assay were shown in Fig. 8. All the four genes of A. thaliana were up-regulated to different degrees when it was treated with QB and analogues. TFQB (8), IQB (21), TQB (22), DHQB (23) and DHTQB (24) showed the abilities inducing MAPKKK18 and RD29A expression equivalent to QB (3) and close to ABA. TFQB (8), IQB (21), TQB (22) and DHQB (23) also displayed the equivalent inducing ability with QB and TFQB for RD298, however, they were slightly weaker for ABF3. TFTQB (25) and iso-QB (26) gave the worst induction activity, strongly related to their bad receptor affinities. These results were highly consistent with their drought resistance and stomatal closure inducing activities.
Quinabactin (QB, 3) is an important non-ABA functional analogue, and it together with its derivative TFQB (8) has great application potential in reducing dehydration and quality deterioration of vegetables during storage (Ma et al., 2018). Herein, we had synthesized several quinabactin analogues by fine-tuning the structure of quinabactin, and discussed the differences of their binding microenvironment with ABA receptors and their effects on plant phenotypes. The results showed that these subtle structural changes had different influences on different physiological processes. Generally speaking, the change of sulfonamide orientation had the greatest influence on the binding of receptor and bioactivity, which was in sharp contrast with the opposite sulfonamide orientation of pyrabactin and quinabactin which had excellent activity. Secondly, very unexpectedly, the thiolation of TFQB (8) led to a sharp decrease in the activity of TFTQB (25), which revealed some forbidden areas of quinabactin structural modification. The molecular docking studies provided a reasonable analysis of these differences. Finally, although quinabactin was not as effective as ABA in conferring drought resistance on plants (Okamoto et al., 2013; Cao et al., 2013), its simple structure and relatively low synthetic cost including its analogues still made them have strong market competitive advantages, especially as drought-resistant inducers. This study provided a constructive insight for the design of quinabactin analogues with better bioactivities, and better drought-resistant inducers were worth looking forward to.
4.1. Instrumentation
1H and 13C NMR spectra were recorded at a Bruker Avance DPX300 spectrometer with TMS as the internal standard. HRMS data were obtained with an ESI-Q-TOF instrument. Thermal images were obtained using VarioCAM HD thermal imager. MST experiment was carried out using a NanoTemper monolith NT.115 instrument. RT-PCR was recorded on Power Wave XS2. Melting points were determined with Yanagimoto MFG melting point apparatus and were uncorrected.
4.2. Synthetic procedures
4.2.1. Synthesis of N-propyl benzolactams (28a, 28b and 32).
The mixture of 100 mL of DMF and 68 mmol of benzolactam (27a, 27b or 31) was cooled to 0℃ under stirring. 6.86 g of 60% sodium hydride was carefully added in batches and stirring was continued at 0℃ for 30 min. 68 mmol of 1-bromopropane was then dropwise added, and the reaction was continued for another 30 min. The ice bath was then removed and the reaction was continued for 2 hours at room temperature. The mixture was poured into crushed ice and extracted with ethyl acetate (3 × 50 mL). The combined organic phase was washed twice with 50 mL of brine, dried overnight with anhydrous sodium sulfate and then filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography (petroleum ether (PE) : ethyl acetate = 10 : 1) to afford pure product.
Data for 28a. Yellow oil, yield 93%. 1H NMR (300 MHz, CDCl3) δ 7.24 (ddd, J = 8.6, 4.8, 0.9 Hz, 2H), 7.01 (td, J = 7.5, 0.9 Hz, 1H), 6.82 (d, J = 7.7 Hz, 1H), 3.66 (t, J = 7.3 Hz, 2H), 3.50 (s, 2H), 1.76–1.64 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H).
Data for 28b. Yellow oil, yield 96%. 1H NMR (300 MHz, CDCl3) δ 7.26–7.17 (m, 1H), 7.15–7.13 (m, 1H), 7.01–6.97 (m, 2H), 3.89 (t, J = 7.3 Hz, 2H), 2.91–2.86 (m, 2H), 2.63 (dd, J = 8.6, 6.1 Hz, 2H), 1.71–1.61 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H).
Data for 32. Yellow oil, yield 95%. 1H NMR (300 MHz, CDCl3) δ 7.51 (d, J = 9.5 Hz, 1H), 7.42 (t, J = 7.4 Hz, 2H), 7.21 (d, J = 8.7 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.56 (d, J = 9.5 Hz, 1H), 4.11 (t, J = 7.3 Hz, 2H), 1.71–1.59 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H).
4.2.2. Synthesis of 5-nitro-1-propylindolin-2-one (29a)
50 mL of concentrated sulfuric acid was cooled to 0℃ and 28 mmol 28a was added under stirring. 31 mmol potassium nitrate was added in batches and the reaction temperature was maintained to not more than 5℃ during addition. The reaction was continued for 4h at this temperature, and then the mixture was poured into ice water and extracted with ethyl acetate (3 × 30 mL). The combined organic phase was washed twice with 30 mL of brine, dried overnight with anhydrous sodium sulfate and then filtered. The filtrate was concentrated in vacuo to give a crude product which provided pure 29a as a yellow solid by column chromatography (PE : EtOAc = 3 : 1), yield 90%, m.p. 97.0–98.0 ℃. 1H NMR (300 MHz, CDCl3) δ 8.26 (dd, J = 8.7, 2.3 Hz, 1H), 8.15 (m, 1H), 6.92 (d, J ༝ 8.7 Hz, 1H), 3.74 (t, J ༝ 7.3 Hz, 2H), 3.64 (s, 2H), 1.80–1.67 (m, 2H), 0.99 (t, J ༝ 7.4 Hz, 3H).
4.2.3. Synthesis of 6-nitro-1-propyl-3,4-dihydroquino-lin-2(1H)-one (29b) and 6-nitro-1-propyl-quinolin-2(1H)-one (33)
50 mL concentrated sulfuric acid was cooled to 0℃ and 26 mmol 28b (or 32) was added under stirring. 10 mL of nitric acid was dropped in and the temperature was maintained not more than 5℃ during the course. The reaction was continued at this temperature for 2 h, and the reaction mixture was then poured into ice water and extracted with ethyl acetate (30 mL × 3). The combined organic phase was washed twice with 30 mL of brine, dried overnight with anhydrous sodium sulfate and then filtered. The filtrate was concentrated in vacuo and the residue was purified by column chromatography (PE: EtOAc = 3: 1).
Data for 29b. Yellow powder, yield 92%, m.p. 103.0-104.0 ℃. 1H NMR (300 MHz, CDCl3) δ 8.15 (dd, J = 9.0, 2.6 Hz, 1H), 8.07 (d, J = 2.5 Hz, 1H), 7.07 (d, J = 9.0 Hz, 1H), 4.02–3.88 (t, J = 7.3 Hz, 2H), 3.08–2.95 (m, 2H), 2.72 (dd, J = 8.8, 6.2 Hz, 2H), 1.67 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H).
Data for 33. Yellow powder, yield 91%, m.p. 106.0–107.0 ℃. 1H NMR (300 MHz, CDCl3) δ 8.48 (d, J = 2.6 Hz, 1H), 8.38 (dd, J = 9.3, 2.6 Hz, 1H), 7.76 (d, J = 9.6 Hz, 1H), 7.43 (d, J = 9.4 Hz, 1H), 6.82 (d, J = 9.5 Hz, 1H), 4.28 (t, J = 7.3 Hz, 2H), 1.85–1.72 (m, 2H), 1.07 (t, J = 7.4 Hz, 3H).
4.2.4. Synthesis of 5-amino-1-propylindolin-2-one (30a), 6-amino-1-propyl-3,4-dihydroquinolin-2(1H)-one (30b) and 6-amino-1-propylquinolin-2(1H)-one (34).
21 mmol of compound 29a (or 29b or 33) and 0.25 g 5% Pd/C were added to a 100 mL round bottom flask, then sealed with a rubber stopper and replaced three times with hydrogen. 30 mL of methanol was added to the reaction flask with a syringe, and the reduction was undergone at room temperature for 3 h. The catalyst was removed by diatomite filtration and the filtrate was concentrated to dryness in vacuo. The residue was purified by column chromatography (PE : EtOAc = 2 : 1) to provide pure product.
Data for 30a. Yellow solid, yield 97%, m.p. 81.0–82.0 ℃. 1H NMR (300 MHz, CDCl3) δ 6.66–6.56 (m, 3H), 3.61 (t, J = 7.5 Hz, 2H), 3.44 (s, 2H), 3.26 (bs, 2H), 1.73–1.61 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H).
Data for 30b. Yellow solid, yield 98%, m.p. 84.0–85.0 ℃. 1H NMR (300 MHz, CDCl3) δ 6.79 (d, J = 8.5 Hz, 1H), 6.58–6.52 (m, 2H), 3.84 (t, J = 7.3 Hz, 2H), 3.59 (s, 2H), 2.76 (dd, J = 8.8, 5.8 Hz, 2H), 2.59 (dd, J = 8.8, 5.8 Hz, 2H), 1.68–1.60 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H).
Data for 34. Yellow solid, yield 98%, m.p. 101.0–102.0 ℃. 1H NMR (300 MHz, CDCl3) δ 7.48 (d, J = 9.4 Hz, 1H), 7.15 (d, J = 9.0 Hz, 1H), 6.95 (dd, J = 8.9, 2.7 Hz, 1H), 6.81 (d, J = 2.7 Hz, 1H), 6.63 (d, J = 9.4 Hz, 1H), 4.19 (t, J = 7.3 Hz, 2H), 3.77 (bs, 2H), 1.80–1.67 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H).
4.2.4. Synthesis of IQB (21) and DHQB (23)
The mixture of 20 mL methylene dichloride (DCE), 10 mmol 30a and 20 mmol of triethylamine was cooled to 0℃. 8 mL DCE solution containing 11 mmol of 4-tolylmethanesulfonyl chloride was dropwise added and stirred for 30 min at 0℃ and then for 2 h at room temperature. 30 mL of water was added and the mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic phase was washed twice with 20 mL of brine and dried with anhydrous sodium sulfate. After evaporating the solvent to dryness in vacuo, the residue was purified by column chromatography (PE : EtOAc = 3 : 1) to give IQB.
Compound QB (3), DHQB (23) were synthesized by the same procedure starting from 30b. While the reaction of 30b with 4-(2,3,5,6-tetrafluoro)tolylmetane sulfonyl chloride in this way would give TFQB (8).
Data for IQB (21). White solid, yield 90%, m.p. 171.8–172.8 ℃. 1H NMR (300 MHz, CDCl3) δ 7.19–7.12 (m, 5H), 7.05 (dd, J = 8.3, 2.2 Hz, 1H), 6.77 (d, J = 8.3 Hz, 1H), 6.59 (s, 1H), 4.26 (s, 2H), 3.67 (t, J = 7.3 Hz, 2H), 3.50 (s, 2H), 2.34 (s, 3H), 1.72–1.67 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 174.36, 142.13, 138.60, 130.91, 130.38, 129.21, 125.65, 125.22, 121.34, 118.87, 108.29, 56.93, 41.40, 35.50, 20.85, 20.42, 11.06. HRMS (m/z) calcd. for C19H22N2O3S [M + H]+ 359.1424, found 359.1418.
Data for QB (3). White solid, yield 91%, m.p. 142.0–143.0 ℃. 1H NMR (300 MHz, CDCl3) δ 7.18–7.14 (m, 4H), 7.02–6.91 (m, 3H), 6.57 (s, 1H), 4.28 (s, 2H), 3.88 (t, J = 7.3 Hz, 2H), 2.88–2.83 (m, 2H), 2.65–2.60 (m, 2H), 2.34 (s, 3H), 1.68–1.63 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 169.53, 138.55, 136.54, 131.39, 130.41, 129.17, 127.64, 125.21, 120.45, 119.51, 115.28, 57.10, 43.41, 31.29, 25.27, 20.85, 20.09, 10.87. HRMS (m/z) calcd. for C20H24N2O3S [M + H]+ 373.1580, found 373.1575.
Data for DHQB (23). Yellowish solid, yield 92%, m.p. 198.0–199.0 ℃. 1H NMR (300 MHz, CDCl3) δ 7.57 (d, J = 9.5 Hz, 1H), 7.36–7.28 (m, 3H), 7.19–7.08 (m, 5H), 6.71 (d, J = 9.5 Hz, 1H), 4.30 (s, 2H), 4.23 (t, J = 7.3 Hz, 2H), 2.31 (s, 3H), 1.81–1.74 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 161.46, 138.69, 138.06, 136.21, 131.07, 130.40, 129.23, 125.03, 123.50, 122.48, 121.23, 119.74, 114.98, 57.32, 43.65, 20.83, 20.55, 11.00. HRMS (m/z) calcd. for C20H22N2O3S [M + H]+ 371.1424, found 371.1419.
Data for TFQB (8). White solid, yield 90%, m.p. 178.0-179.0 ℃. 1H NMR (300 MHz, DMSO-d6) δ 10.05 (s, 1H), 7.06–7.02 (m, 3H), 4.56 (s, 2H), 3.79 (t, J = 7.3 Hz, 2H), 2.79–2.77 (m, 2H), 2.76–2.75 (m, 2H), 2.22 (s, 3H), 1.51–1.46 (m, 2H), 0.85 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ 168.89, 146.12 (m), 142.92 (m), 135.78, 132.48, 127.34, 119.34, 118.68, 116.98 (t, J = 19.4 Hz), 115.67, 106.82 (t, J = 17.7 Hz), 46.08, 42.64, 31.34, 25.09, 20.10, 11.10, 7.53. HRMS (m/z) calcd. for C20H20F4N2O3S [M + H]+ 445.1204, found. 445.1199
4.2.5. Synthesis of TQB (22), DHTQB (24) and TFTQB (25)
A mixture of 15 mL THF, 2.68 mmol quinabactin and 1.47 mmol phosphorus pentasulfide were stirred at 60℃ for 2 h. After cooling to room temperature, the mixture was filtered with diatomite and the filtrate was concentrated to dryness, the residue was purified to give TQB by column chromatography (PE : EtOAc = 6 : 1). DHTQB and TFTQB were prepared by same procedures.
Data for TQB (22). Yellow solid, yield 89%, m.p. 175.0–176.0 ℃. 1H NMR (300 MHz, CDCl3) δ 7.12–7.05 (m, 4H), 7.09–6.85 (m, 3H), 6.56 (s, 1H), 4.39 (t, J = 7.5 Hz, 2H), 4.22 (s, 2H), 3.12–3.07 (m, 2H), 2.69–2.65 (m, 2H), 2.27 (s, 3H), 1.82–1.74 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 198.32, 138.09, 135.23, 132.30, 129.79, 129.71, 128.59, 124.26, 118.75, 117.99, 116.26, 56.75, 50.67, 41.21, 24.02, 20.21, 18.58, 10.04. HRMS (m/z) calcd. for C20H24N2O2S2 [M-H]− 387.1206, found 387.1221.
Data for DHTQB (24). Yellow solid, yield 75%, m.p. 227.0-228.0 ℃. 1H NMR (300 MHz, DMSO-d6) δ 7.57 (d, J = 9.1 Hz, 1H), 7.40 (d, J = 8.9 Hz, 1H), 7.28 (dt, J = 12.2, 6.0 Hz, 3H), 7.12–7.04 (m, 4H), 6.82 (s, 1H), 4.78 (t, J = 7.3 Hz, 2H), 4.28 (s, 2H), 2.25 (s, 3H), 1.86–1.78 (m, 2H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 198.49, 138.22, 136.31, 133.73, 132.20, 129.82, 129.74, 128.63, 124.39, 124.15, 122.64, 117.64, 115.89, 57.11, 50.37, 20.17, 18.83, 10.07. HRMS (m/z) calcd. for C20H22N2O2S2 [M + H]+ 387.1195, found 387.1191.
Data for TFTQB (24). Yellow solid, yield 87%, m.p. 207.0-208.0 ℃. 1H NMR (300 MHz, CDCl3) δ 7.15–7.05 (m, 3H), 6.78 (s, 1H), 4.52 (s, 2H), 4.49–4.43 (m, 2H), 3.18 (dd, J = 8.4, 5.9 Hz, 2H), 2.78 (dd, J = 8.3, 6.0 Hz, 2H), 2.26 (s, 3H), 1.89–1.78 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 198.50, 144.91 (m), 142.19 (m), 135.64, 131.61, 129.84, 120.99 (t, J = 20.9 Hz), 118.67, 117.98, 116.28, 114.65 (t, J = 16.6 Hz), 50.65, 45.44, 41.21, 24.03, 18.56, 10.01, 6.78. HRMS (m/z) calcd. for C20H20F4N2O2S2 [M + H]+ 461.0975, found 461.0968.
4.2.6. Synthesis of iso-QB (26)
10 mL of chlorosulfonic acid was slowly drop to 10 mmol of 28b below 0℃ under stirring. The obtained mixture was stirred for two hours and then poured into 30 mL ice water, extracted with ethyl acetate (3 × 20 mL). The combined organic phases were washed twice with 20 mL brine, and dried with anhydrous sodium sulfate. After evaporating the solvent in vacuo, the crude product 35 was obtained and was directly used for the next reaction without further purification.
A solution of 2 mmol of 35 dissolved in 2 mL DCE was dropped into a mixture of 2 mmol 4-methylbenzylamine, 8 mL DCE and 4 mmol triethylamine at 0℃ under stirring. The reaction was continued for 30 min at 0℃ and then for 4 h at room temperature. Poured the reaction mixture into ice water and extracted with ethyl acetate (3 × 10 mL). The combined organic phases were washed twice with 10 mL brine and dried with anhydrous sodium sulfate. After evaporating the solvent in vacuo, the residue was purified by column chromatography (PE : EtOAc = 2 : 1) to give 26 as white solid with 90% yield, m.p. 101.5-102.5 ℃. 1H NMR (300 MHz, CDCl3) δ 7.72 (dd, J = 8.6, 2.2 Hz, 1H), 7.60 (d, J = 2.2 Hz, 1H), 7.09–7.00 (m, 5H), 5.22 (t, J = 6.2 Hz, 1H), 4.09 (d, J = 6.2 Hz, 2H), 3.89 (t, J = 7.3 Hz, 2H), 2.88–2.85 (m, 2H), 2.66–2.61 (m, 2H), 2.27 (s, 3H), 1.66–1.61 (m, 2H), 0.96 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 169.57, 142.92, 137.16, 133.29, 133.01, 128.88, 127.54, 126.77, 126.61, 126.57, 114.50, 46.65, 43.47, 30.97, 24.99, 20.71, 19.96, 10.82. HRMS (m/z) calcd for C20H24N2O3S [M + H]+ 373.1580, found 373.1575.
4.3. Bioassays
4.3.1. A. thaliana and Lettuce seed germination assay
This assay was conducted according to the method we described earlier (Che et al., 2020).
4.3.2. Seedling growth inhibition
Plump rice seeds with same size were sterilized with 70% ethanol for 10 min, and then rinsed with ddH2O for 6–7 times. They were immersed in distilled water and swelled to just budding. Then they were transferred to sterilized test tubes which contains 2 mL tested agent with different concentration in each one. After sealing with sealing membranes, they were placed in an incubator for cultivation under 28℃, 16 h illumination and 25℃, 8 h darkness alternatively for one week. The lengths of the second blades were counted. Each experiment was repeated three times.
A. thaliana seeds were sterilized with 70% ethanol for 10 min, and then rinsed with ddH2O for 6–7 times. 25 seeds were seeded on a Petri dish with 1/2 MS solid culture medium. After drying, they were sealed and placed in a refrigerator at 4℃ for 3 days to vernalize and then transferred to an incubator for cultivation of 3 days under 25℃, 16 h illumination and 22℃, 8 h darkness alternatively. The 3-day-old seedlings were transferred to a 1/2 MS solid culture medium containing 25 µM of tested agent and placed in a light incubator for vertical culture. One week later, the lengths of primary roots and the fresh weight of leaves of Arabidopsis seedlings were measured. Each treatment was repeated three times.
4.3.3 Drought-resistance induction
Plump wheat seeds were sterilized with 70% ethanol for 10 min, and then rinsed with ddH2O for 6–7 times. They were immersed in distilled water and swelled to just budding and then planted in sterilized nutrient soil and cultivated in a light incubator under 25℃, 14 h illumination and 20℃, 10 h darkness alternatively for two weeks. The water was cut-off from the 15th day and each pot was sprayed with 10 mL of 25 µM tested agent. One week later, the wilting appearance of leaves were photographed and the fresh weight of aboveground parts and soil moisture contents were measured. The soil moisture contents were measured by the oven drying method: the soil samples were taken into the oven at 120 ℃ and dried to constant weight, which was the weight of the dried soil. On this basis, the percentage (%) of water weight (evaporation loss) was calculated to obtain the soil moisture content. Each experiment was repeated three times.
Soybean seeds with full grains were sterilized with 70% ethanol for 10 min, and then rinsed with ddH2O for 6–7 times. They were soaked in distilled water and swelled to just budding and then planted in sterilized nutrient soil and cultivated in a light incubator under 25℃, 16 h illumination and 20℃, 8 h darkness alternatively for two weeks. The water was cut-off from the 15th day and each pot was sprayed with 10 mL of 25 µM tested agent every 2 days. One week later, the wilting appearance of leaves were photographed and the soil moisture contents were measured. Each experiment was repeated three times.
A. thaliana seeds were sterilized with 70% ethanol for 10 min, and then rinsed with ddH2O for 6–7 times. They were seeded on a Petri dish with 1/2 MS solid culture medium. After drying, they were sealed and placed in a refrigerator at 4℃ for 3 days to vernalize and then transferred to an incubator for cultivation of 5 days under 25℃, 16 h illumination and 22℃, 8 h darkness alternatively. The 5-day-old seedlings were planted in sterilized nutrient soil to grow in greenhouse for two weeks. The water was cut-off from the 15th day and each pot was sprayed with 10 mL of 25 µM tested agent every 3 days. Ten days later, the wilting appearance of leaves were photographed and the soil moisture contents were measured. Each experiment was repeated three times.
4.3.4. A. thaliana stomatal closure assay
This assay was conducted by the method we described earlier (Che et al., 2020).
4.3.5. Receptor binding and phosphatase HAB1 inhibition assay
These assays were conducted by the method we described earlier (Che et al., 2020).
4.3.6. RT-PCR assay (Okamoto et al., 2013)
A. thaliana seeds were sterilized with 70% ethanol for 10 min, and then rinsed with ddH2O for 6–7 times. They were seeded on a Petri dish with 1/2 MS solid culture medium. After drying, they were sealed and placed in a refrigerator at 4℃ for 3 days to vernalize and then transferred to an incubator for cultivation of 10 days under 25℃, 16 h illumination and 22℃, 8 h darkness alternatively. These 10-day-old seedlings were treated with 25 µM test agents for 6 h and total RNA was extracted by TRIzol (Invitrogen) method, and contaminated DNA was removed by RNase-free DNase (Qiagen) before RT-PCR. For quantitative RT-PCR, total RNA was reverse transcribed with TransScript RT kit (Invitrogen). The quantitative RT-PCR assay was performed in CFX96 real-time system (BIORAD) according to the two-step protocol of SYBR Premix Ex Taq TM II kit (TaKaRa). Each experiment was repeated three times, and ACT2 was used as internal control in quantitative RT-PCR.
4.4. Molecular docking
Primarily, the 3D structural models were minimized with the staged minimization option on SYBYL (Tripos force field) (Clark et al., 1989), using the Powell optimization method with Gasteiger-Huckel point charge (Fletcher and Powell, 1963). The energy gradient convergence value was set to 0.005 kcal/(mol⋅Å) and the max iterations was 1000 times. Then, the compounds were docked with PYL2(PDB_ID: 3KDI) and PYL5(PDB_ID: 4JDL) using Surflex-Dock (Jain, 2003).
Declaration of competing interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 22077136).
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://
Author contribution statement
Xiaobin Li and Chuanliang Che designed the project and performed the experiments. Huizhe Lu analyzed the data. Xiaobin Li and Chuanliang Che wrote the manuscript. Yong Xu, Xianjun Tang, Yanjun Xu and Xueqin Zhang helped conduct the experiments. Yumei Xiao, Jia-Qi Li and Zhaohai Qin revised the manuscript. All authors performed a systematic search of bibliography about the topic, wrote and revised the manuscript.
- Ban, T., Shiozaki, S., Ogata, T., Horiuchi, S., 2000, Effects of abscisic acid and shading treatments on the levels of anthocyanin and resveratrol in skin of Kyoho grape berry. Acta Hortic. 514, 83-90.
- Benson, C.L., Kepka, M., Wunschel, C., Rajagopalan, N., Nelson, K.M., Christmann, A., Abrams, S.R., Grill, E., Loewen, M.C., 2015, Abscisic acid analogs as chemical probes for dissection of abscisic acid responses in Arabidopsis thaliana. Phytochemistry. 113, 96-107.
- Cao, M., Liu, X., Zhang, Y., Xue, X.Q., Zhou, X.E., Melcher, K., Gao, P., Wang, F.X., Zeng, L., Zhao, Y., Zhao, Y., Deng, P., Zhong, D.F., Zhu, J.K., Xu, H.E., Xu, Y., 2013, An ABA-mimicking ligand that reduces water loss and promotes drought resistance in plants. Cell Res. 23, 1043-1054.
- Cao, M.J., Zhang, Y.L., Liu, X., Huang, H., Zhou, X.E., Wang, W.L., Zeng, A., Zhao, C.Z., Si, T., Du, J.M., Wu, W.W., Wang, F.X., Xu, H.E., Zhu, J.K., 2017, Combining chemical and genetic approaches to increase drought resistance in plants. Nat. Commun. 8, 1183.
- Che, C. L., Zeng, Y.J., Xu, Y.F., Lu, H.Z., Xu, Y.J., Zhang, X.Q., Xiao, Y.M., Li, J.Q., Qin, Z.H., 2020, APAn, a class of ABA receptor agonism/antagonism switching probes, J. Agric. Food Chem. 68, 8524−8534.
- Clark, M., Cramer, R.D., Vanopdenbosch, N., 1989, Validation of the general-purpose Tripos 5.2 Force-Field. J. Comput. Chem. 10, 982-1012.
- Cutler, S.R., Rodriguez, P.L., Finkelstein, R.R., Abrams, S.R., 2010, Abscisic acid: Emergence of a core signaling network. Annu. Rev. Plant Biol. 61, 651–679.
- Elzinga, D., Sternburg, E., Sabbadin, D., Bartsch, M., Park, S.Y., Vaidya, A., Mosquna, A., Kaundal, A., Wendeborn, S., Lachia, M., Karginov, F.V., Cutler, S.R., 2019, Defining and Exploiting Hypersensitivity Hotspots to Facilitate Abscisic Acid Agonist Optimization, ACS Chem. Biol. 14, 332−336
- Fletcher, R., Powell, M.J.D., 1963, A rapidly convergent descent method for minimization. Comput. J. 6, 163-168.
- Han, X.Q., Jiang, L., Che, C.L., Wan, C., Lu, H.Z., Xiao, Y.M., Xu, Y.J., Chen, Z.Z., Qin, Z.H., 2017, Design and functional characterization of a novel abscisic acid analog. Sci. Rep. 7, 43863.
- Kim, B.T., Min, Y.K., Asami, T., Park, N.K., Jeong, I.H., Cho, K.Y., Yoshida, S., 1995, Synthesis and biological activities of new fluorinated abscisic acid. Bioorg. Med. Chem. Lett. 5, 275-278.
- Jain, A.N., 2003, Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 46, 499-511.
- Jones, L.H., 2020, Dehydroamino acid chemical biology: an example of functional group interconversion on proteins, RSC Chem. Biol. 1, 298-304.
- Ma, D.Y., Xu, Y., Zhang, Z.Q., Li, B.Q., Chen, T., Tian, S. P., 2018, Efficacy of ABA-mimicking ligands in controlling water loss and maintaining antioxidative capacity of Spinacia oleracea. J. Agric. Food Chem. 66, 13397−13404.
- Ma, Y., Szostkiewicz, I., Korte, A., Moes, D., Yang, Y., Christmann, A., Grill, E., 2009, Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science. 324, 1064-1068.
- McCarty, D.R., 1995, Genetic-control and integration of maturation and germination pathways in seed development. Annu. Rev. Plant Biol. 46, 71–93.
- Munns, R., James, R.A., Lauchli, A., 2006, Approaches to increasing the salt tolerance of wheat and other cereals. J. Exp. Bot. 57, 1025-1043.
- Nyangulu, J.M., Nelson, K.M., Rose, P.A., Gai, Y.Z., Loewen, M., Lougheed, B., Quail, J.W., Cutler, A.J., Abrams, S.R., 2006, Synthesis and biological activity of tetralone abscisic acid analogues. Org. Biomol. Chem. 4, 1400-1412.
- Ohkuma, K., Lyon, J.L., Addicott, F.T., Smith, O.E., 1963, Abscisin II, an Abscission-Accelerating Substance from Young Cotton Fruit. Science. 142, 1592-1593.
- Okamoto, M., Peterson, F.C., Defries, A., Park, S.Y., Endo, A., Nambara, E., Volkmanc, B.F., Cutler, S.R., 2013, Activation of dimeric ABA receptors elicits guard cell closure, ABA-regulated gene expression, and drought tolerance. Proc. Natl. Acad. Sci. USA. 110, 12132-12137.
- Oritani, T., Kiyota, H., 2003, Biosynthesis and metabolism of abscisic acid and related compounds. Nat. Prod. Rep. 20, 414-425.
- Park, S.Y., Fung, P., Nishimura, N., Jensen, D.R., Fujii, H., Zhao, Y., Lumba, S., Santiago, J., Rodrigues, A., Chow, T.F.F., Alfred, S.E., Bonetta, D., Finkelstein, R., Provart, N.J., Desveaux, D., Rodriguez, P.L., McCourt, P., Zhu, J.K., Schroeder, J.I., Volkman, B.F., Cutler, S.R., 2009, Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science. 324, 1068-1071.
- Plancher, B., 1979, Notes on the isomerization of abscisic acid by irradiation with UV light. Gartenbauwissenschaft. 44, 184-191.
- Teng, K.Q., Li, J.Z., Liu, L., Han, Y.C., Du, Y.X., Zhang, J., Sun, H.Z., Zhao, Q.Z., 2014, Exogenous ABA induces drought tolerance in upland rice: the role of chloroplast and ABA biosynthesis-related gene expression on photosystem II during PEG stress, Acta Physiol Plant. 36, 2219–2227.
- Valluru, R., Link, J., Claupein, W., 2012, Consequences of early chilling stress in two Triticum species: plastic responses and adaptive significance. Plant Biol. 14, 641-651.
- Vaidya, A.S., Peterson, F.C., Yarmolinsky, D., Merilo, E., Verstraeten, I., Park, S.Y., Elzinga, D., Kaundal, A., Helander, J., Juste, J.L., Otani, M., Wu, K., Jensen, D.R., Kollist, H., Volkman, B.F., Cutler, S.R., 2017, A rationally designed agonist defines subfamily IIIA abscisic acid receptors as critical targets for manipulating transpiration. ACS Chem. Biol. 12, 2842-2848.
- Vaidya, A.S., Helander, J., Peterson, F.C., Elzinga, D., Dejonghe, W., Kaundal, A., Park, S.Y., Xing, Z.N., Mega, R., Takeuchi, J., Khanderahoo, B., Bishay, S., Volkman, B.F., Todoroki, Y., Okamoto, M., Cutler, S.R., 2019, Dynamic control of plant water use using designed ABA receptor agonists. Science. 366, eaaw8848.
- Yamaguchi-Shinozaki, K., Shinozaki, K., 2006, Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu. Rev. Plant Biol. 57, 781–803.
- Yoshida, K., Kondoh, Y., Nakano, T., Bolortuya, B., Kawabata, S., Iwahashi, F., Nagano, E., Osada, H., 2021, New abscisic acid derivatives revealed adequate regulation of stomatal, transcriptional, and developmental responses to conquer drought. ACS Chem. Biol. 16, 1566−1575.
- Zeevaart, J.A.D., Creelman, R.A., 1998, Metabolism and physiology of abscisic acid. Annu. Rev. Plant Biol. 39, 439–473.
Schemes are available in the Supplementary Files section.
No competing interests reported.
- Scheme1.png
Scheme 1. From lead molecules to modified compounds
- Scheme2.png
Scheme 2. Synthetic pathway of QB (3), TFQB (8) and their analogues with saturated lactam ring
- Scheme3.png
Scheme 3. Synthetic pathway of QB (3) analogues with unsaturated lactam ring
- Scheme4.png
Scheme 4. Synthetic pathway of QB (3) analogue with a reversed sulphonamide group
- SupportingInformation.docx
{kind=link}
{kind=link}
{kind=link}
{kind=link}