2.1 Traditional reaction mechanisms
We model the OER over a (2x1) IrO2(110) surface by DFT calculations; all computational details can be found in section 1 of the SI. The stoichiometric (2x1) IrO2(110) surface, abbreviated
2Obr + 2*cus (cf. SI, Figure S1), contains two dissimilar Ir surface atoms with a different chemical environment, namely ‘cus’ and ‘bridge’ sites41. There is a consensus in the literature that the cus sites are the catalytically active centers for the OER or other surface reactions whereas the bridge sites are mainly spectators. Note that the bridge sites can still be involved in the OER, as oxygen atoms at the Ir bridge site can serve as a Brønsted base by accepting a proton during the elementary steps of the OER (cf. SI, section 3.2 and 3.3).42,43
To gain insight into the surface configuration of IrO2 during the OER, we apply the concept of surface Pourbaix diagrams to identify thermodynamically stable structure under anodic conditions.44–47 A detailed discussion of the Pourbaix approach is provided in section 2 of the SI. Figure 1a indicates the DFT-based Pourbaix diagram of a single-crystalline IrO2(110) electrode as a function of applied electrode potential and pH.
Figure 1. a) Pourbaix diagram for IrO2(110), indicating the thermodynamically most stable surface phase as a function of applied electrode potential, U, and pH. The white dotted line represents the OER equilibrium potential, U0OER = 1.23 V vs. reversible hydrogen electrode (RHE). While a partly hydroxylated IrO2(110) surface is observed at electrode potentials exceeding the OER equilibrium potential, we consider four different surface motifs for our mechanistic studies due to their comparable energetics under OER conditions: b) fully hydroxylated surface (2OHbr + 2 *cus-OHot), c) partly hydroxylated surface (2Obr + 2*cus-OHot), d) fully oxygen-covered surface (2Obr + 2*cus-Oot), and e) partly OOH-terminated surface (2Obr + 1*cus-OOHot 1*cus-Oot).
Figure 1a reveals that depending on the applied electrode potential, different hydroxylated, oxygen-covered, and OOH-covered surfaces are energetically preferred. To this end, we select four different surface configurations (cf. Figure 1b-e) with comparable energetics under typical OER conditions (U > 1.23 V vs. RHE) as the starting point for our mechanistic analyses. Initially, we consider five different reaction mechanisms, namely the mononuclear, bifunctional I, bifunctional II, oxide, and binuclear descriptions (cf. SI, section 3).48–51 These pathways are summarized in Fig. 2 by a network of elementary steps using the example of the fully oxygen-covered surface (cf. Figure 1d).
To assess the electrocatalytic activity of the different mechanisms over the various surface configurations, we employ the descriptor Gmax(U), which relies on the notion of a span model:52,53 Gmax(U) indicates the largest free-energy span from the intermediate with the smallest to the intermediate with the highest free energy at a given electrode potential in the free-energy landscape. The peculiarity of this descriptor refers to the fact that it contains a measure for sensitivity54,55, based on the benchmarking with experimentally obtained transition-state free energies:22,56 only if the Gmax(U) values of two mechanisms differ by at least 0.20 eV, we infer that the mechanism with the smaller Gmax(U) value is unambiguously preferred. This allows the screening of mechanistic pathways of proton-coupled electron transfer steps on the level of thermodynamic considerations, focusing on the free energies of the intermediate species. We note that the application of Gmax(U) as the activity descriptor in our analysis does not cause a significant loss in accuracy as even state-of-the-art transition state calculations for proton-coupled electron transfer steps under constant potential contain error bars of about 0.15 eV.33,57,58 To this end, we conclude that it is beneficial to use the thermodynamic evaluation with a slightly larger uncertainty than the kinetic picture, as only this simplification allows us to conduct a comprehensive study of the various pathways (cf. Figure 2) and surface configurations (cf. Figure 1b-e) for the OER over IrO2(110).
In section 4 of the SI, we discuss the OER mechanisms over the fully hydroxylated IrO2(110) surface – 2OHbr + 2*cus-OHot – by the construction of free-energy diagrams. Table 1 summarizes the results of these mechanistic investigations, pinpointing that two mechanisms are competing under OER conditions of U = 1.53 V vs. RHE: both bifunctional descriptions reveal identical electrocatalytic activity in the approximation of Gmax(U) whereas the other pathways can be fairly ruled out because of their larger values of the activity measure Gmax(U). Please note that the oxide mechanism cannot take place for the fully hydroxylated IrO2(110) surface due to the instability of a reaction intermediate containing the *OOH adsorbate (cf. SI, section 4.4). We note that the observation of the bifunctional description under OER conditions coincides with the previous works of Goddard and coworkers or Jones and coworkers.20,21
For the partly hydroxylated IrO2(110) surface – 2Obr + 2*cus-OHot – we observe that four mechanistic descriptions compete under OER conditions (cf. Table 2). While the oxide mechanism59 is energetically preferred due to the smallest value of the descriptor Gmax(U), the mononuclear and bifunctional descriptions cannot be excluded due to their comparable electrocatalytic activity. Only the binuclear mechanism can be clearly ruled out because of the large energy penalty for the formation of gaseous oxygen by a chemical reaction step. It is relevant to point out that the oxide mechanism as the operating pathway is in line with the previous work by Binninger and Doublet.24 For further information on the OER mechanisms over the partly fully hydroxylated IrO2(110) surface, we refer the reader to section 5 of the SI.
Table 1
Energetic assessment of the various mechanistic pathways (cf. Figure 2) for the OER over a fully hydroxylated IrO2(110) surface (cf. Figure 1b) by the descriptor Gmax(U) at U = 1.23 V and 1.53 V vs. RHE. For both potential conditions, the limiting free-energy span in the approximation of Gmax(U) is indicated. Further details are provided in section 4 of the SI.
| Mechanisms | U = 1.23 V | U = 1.53 V |
| | Gmax(U) (eV) | Free-energy span | Gmax(U) (eV) | Free-energy span |
| Mononuclear mechanism | 1.76 | *cus-OHot → *cus + O2 | 0.86 | *cus-OHot → *cus + O2 |
| Bifunctional I mechanism | 1.47 | *cus-OHot + Obr → *cus + Obr + O2 | 0.57 | *cus-OHot + Obr → *cus + Obr + O2 |
| Bifunctional II mechanism | 1.47 | *cus-OHot + Obr → *cus + Obr + O2 | 0.57 | *cus-OHot + Obr → *cus + Obr + O2 |
| Oxide mechanism | - | - | - | - |
| Binuclear mechanism | 3.46 | *cus-OHot + *cus-OHot → *cus + *cus + O2 | 2.86 | *cus-OHot + *cus-OHot → *cus + *cus + O2 |
Table 2
Energetic assessment of the various mechanistic pathways (cf. Figure 2) for the OER over a partly hydroxylated IrO2(110) surface (cf. Figure 1c) by the descriptor Gmax(U) at U = 1.23 V and 1.53 V vs. RHE. For both potential conditions, the limiting free-energy span in the approximation of Gmax(U) is indicated. Further details are provided in section 5 of the SI.
| Mechanisms | U = 1.23 V | U = 1.53 V |
| | Gmax(U) (eV) | Free-energy span | Gmax(U) (eV) | Free-energy span |
| Mononuclear mechanism | 1.45 | *cus-OHot → *cus + O2 | 0.75 | *cus-OOHot → *cus + O2 |
| Bifunctional – I mechanism | 1.45 | *cus-OHot + Obr → *cus + Obr + O2 | 0.68 | *cus-OOot + OHbr → *cus + Obr + O2 |
| Bifunctional – II mechanism | 1.45 | *cus-OHot + Obr → *cus + Obr + O2 | 0.75 | *cus-OOHot + Obr → *cus + Obr + O2 |
| Oxide mechanism | 1.07 | *cus-Oot + *cus-Oot → *cus-OOHot + *cus-OOHot | 0.66 | *cus-OOHot + *cus-Oot → *cus-OOHot + *cus-OOHot |
| Binuclear mechanism | 2.78 | *cus-OHot + *cus-OHot → *cus + *cus + O2 | 2.18 | *cus-OHot + *cus-OHot → *cus + *cus + O2 |
It is noteworthy that the limiting span for the four identified pathways of the partly hydroxylated IrO2(110) surface are different to a large extent compared to the case of the fully hydroxylated surface (cf. Table 1). Table 2 indicates that under OER conditions (U = 1.53 V vs. RHE), the decomposition of the *OOH or *OO adsorbates rather than their formation governs the rate in the approximation of Gmax(U). This finding agrees with the suggestion of Exner and Over on the limiting step in the OER over IrO2(110).22
A similar situation is encountered for the fully oxygen-covered IrO2(110) surface – 2Obr + 2*cus-Oot – where the mononuclear and bifunctional descriptions compete under OER conditions. While an extended discussion is provided in section 6 of the SI, Table 3 illustrates that the limiting span for these pathways comprises the decomposition of the *OOH intermediate.
The Pourbaix diagram in Fig. 1 illustrates that at applied electrode potentials exceeding 1.58 V vs. RHE, the *OOH adsorbate becomes thermodynamically stable on the electrode surface. To this end, we study the different mechanistic pathways over the partly OOH-covered IrO2(110) surface – 2Obr + 1*cus-OOHot 1*cus-Oot – and observe that the bifunctional and oxide descriptions are preferred under OER conditions (cf. Table 4). For the OOH-covered surface, also the formation of the *OOH adsorbate can govern the activity descriptor Gmax(U) in case of the oxide pathway, which is in line with the proposal of Rossmeisl and coworkers on the limiting reaction step.16,17,51 Please note that the binuclear mechanism cannot be operative for the partly OOH-covered IrO2(110) surface as further explained in section 7 of the SI, where free-energy diagrams along the reaction coordinate are provided for all the mechanistic descriptions.
In summary, it can be concluded that the different reports on the limiting reaction step and mechanism in the OER over IrO2(110) in the literature are essentially reproduced when considering that different surface configurations are available on the electrode surface under reaction conditions. When comparing the intrinsic activity of these surface configurations in the approximation of Gmax(U), we observe that the fully oxygen-covered surface (cf. Table 3) is the most active phase at U = 1.53 V vs. RHE; however, the fully hydroxylated (cf. Table 1) and partly OOH-covered (cf. Table 1) surfaces cannot be excluded as Gmax(U) deviates less than 0.20 eV compared to the fully oxygen-covered configuration. This finding suggests that not a single mechanism or a single reaction step is governing the OER over IrO2(110), but rather a variety of different steps and mechanisms control the rate of this reaction.48 In the following section though, we demonstrate that none of the above mechanistic descriptions is operative for IrO2(110) under OER conditions due to the necessity of considering Walden-type pathways in the analysis.39,40
Table 3
Energetic assessment of the various mechanistic pathways (cf. Figure 2) for the OER over a fully oxygen-covered IrO2(110) surface (cf. Figure 1d) by the descriptor Gmax(U) at U = 1.23 V and 1.53 V vs. RHE. For both potential conditions, the limiting free-energy span in the approximation of Gmax(U) is indicated. Further details are provided in section 5 of the SI.
| Mechanisms | U = 1.23 V | U = 1.53 V |
| | Gmax(U) (eV) | Free-energy span | Gmax(U) (eV) | Free-energy span |
| Mononuclear mechanism | 1.20 | *cus-OHot → *cus + O2 | 0.41 | *cus-OOHot → *cus + O2 |
| Bifunctional – I mechanism | 1.21 | *cus-OHot + Obr → *cus + Obr + O2 | 0.38 | *cus-OOot + OHbr → *cus + Obr + O2 |
| Bifunctional – II mechanism | 1.21 | *cus-OHot + Obr → *cus + Obr + O2 | 0.39 | *cus-OOHot + Obr → *cus + Obr + O2 |
| Oxide mechanism | 1.07 | *cus-Oot + *cus-Oot → *cus-OOHot + *cus-OOHot | 0.66 | *cus-OOHot + *cus-Oot → *cus-OOHot + *cus-OOHot |
| Binuclear mechanism | 2.78 | *cus-OHot + *cus-OHot → *cus + *cus + O2 | 2.18 | *cus-OHot + *cus-OHot → *cus + *cus + O2 |
Table 4
Energetic assessment of the various mechanistic pathways (cf. Figure 2) for the OER over a partly *OOH-covered IrO2(110) surface (cf. Figure 1e) by the descriptor Gmax(U) at U = 1.23 V and 1.53 V vs. RHE. For both potential conditions, the limiting free-energy span in the approximation of Gmax(U) is indicated. Further details are provided in section 5 of the SI.
| Mechanisms | U = 1.23 V | U = 1.53 V |
| | Gmax(U) (eV) | Free-energy span | Gmax(U) (eV) | Free-energy span |
| Mononuclear mechanism | 1.61 | *cus-OHot → *cus-OOHot | 1.01 | *cus-OHot → *cus-OOHot |
| Bifunctional – I mechanism | 1.46 | *cus-OHot + Obr → *cus + Obr + O2 | 0.56 | *cus-OHot + Obr → *cus + Obr + O2 |
| Bifunctional – II mechanism | 1.61 | *cus-OHot + Obr → *cus-OOHot + Obr | 1.01 | *cus-OHot + Obr → *cus-OOHot + Obr |
| Oxide mechanism | 1.07 | *cus-Oot + *cus-Oot → *cus-OOHot + *cus-OOHot | 0.66 | *cus-OOHot + *cus-Oot → *cus-OOHot + *cus-OOHot |
| Binuclear mechanism | - | - | - | - |
2.2 Walden-type mechanisms
All mechanistic pathways summarized in the network of Fig. 2 rely on the notion that product formation is accompanied with the restoration of the catalytically active Ir cus site, *cus, on the IrO2(110) surface (cf. structure (1) in Fig. 2). Yet, removal of the product O2 and adsorption of the reactant H2O can also proceed simultaneously so that correspondingly, the vacant *cus site is no longer observed in the catalytic cycle. Mechanisms that follow these lines are called Walden pathways,39 and Fig. 3 provides a sketch of the mononuclear- and bifunctional-Walden OER mechanisms. The elementary steps for both mechanistic descriptions are compiled in section 8 of the SI. Table 5 summarizes the mechanistic analysis of the Walden-type mechanisms over the fully hydroxylated, partly hydroxylated, fully oxygen-covered, and partly OOH-covered IrO2(110) surfaces (cf. Figure 1b-e). Free-energy diagrams for the Walden pathways over the four surface configurations are provided in sections 9–12 of the SI.
Table 5 indicates that the electrocatalytic activity in the approximation of Gmax(U) is on the order of 0.12–0.22 eV at U = 1.53 V vs. RHE, except for the partly *OOH-covered surface that reveals Gmax(U) = 0.35 eV. This translates to a reduction of about 0.3–0.6 eV compared to the traditional OER mechanisms (cf. Tables 1–4). Therefore, we arrive at the intriguing finding that the OER on IrO2(110) is governed by Walden-type pathways rather than by any of the previously assumed reaction mechanisms, and this conclusion can be rendered in an unbiased manner by the descriptor Gmax(U) as the free-energy difference between the different pathways clearly exceeds the threshold of 0.20 eV.33
Table 5
Energetic assessment of the Walden pathways (cf. Figure 3) for the OER over different surface motifs of a single-crystalline IrO2(110) electrode (cf. Figure 1b-e) by the descriptor Gmax(U) at U = 1.23 V and 1.53 V vs. RHE. For both potential conditions, the limiting free-energy span in the approximation of Gmax(U) is indicated. Further details are provided in sections 9–12 of the supplemental.
| Mechanisms | U = 1.23 V | U = 1.53 V |
| Gmax(U) (eV) | Free-energy span | Gmax(U) (eV) | Free-energy span |
| Fully hydroxylated IrO2(110) surface |
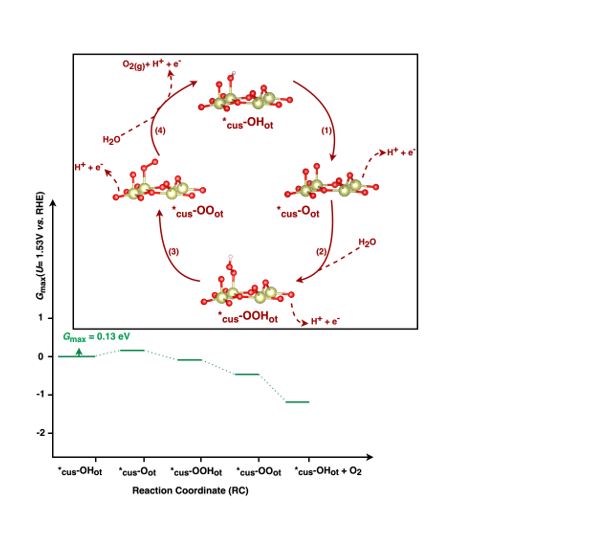
| Mononuclear-Walden | 0.63 | *cus-OHot → *cus-OOHot | 0.12 | *cus-OHot → *cus-Oot |
| Bifunctional-Walden | 0.62 | *cus-OHot + Obr → *cus-OOot + OHbr | 0.13 | *cus-OHot + Obr → *cus-Oot + Obr |
| Partly hydroxylated IrO2(110) surface |
| Mononuclear-Walden | 0.52 | *cus-OHot → *cus-Oot | 0.22 | *cus-OHot → *cus-Oot |
| Bifunctional-Walden | 0.52 | *cus-OHot + Obr → *cus-Oot + Obr | 0.22 | *cus-OHot + Obr → *cus-Oot + Obr |
| Fully oxygen-covered IrO2(110) surface |
| Mononuclear-Walden | 0.53 | *cus-OHot → *cus-OOHot | 0.13 | *cus-OHot → *cus-Oot |
| Bifunctional-Walden | 0.52 | *cus-OHot + Obr → *cus-OOot + OHbr | 0.13 | *cus-OHot + Obr → *cus-Oot + Obr |
| Partly OOH-covered IrO2(110) surface |
| Mononuclear-Walden | 1.61 | *cus-OHot → *cus-OOHot | 1.01 | *cus-OHot → *cus-OOHot |
| Bifunctional-Walden | 1.09 | *cus-OHot + Obr → *cus-OOot + Obr | 0.35 | *cus-OHot + Obr → *cus-Oot + Obr |
Our extended mechanistic analysis also reveals that there is a change in the limiting free-energy span in the Walden-type pathways (cf. Table 5) compared to the traditional mechanisms (cf. Tables 1–4). While the latter are mainly governed by the decomposition of the *OOH adsorbate, the Walden pathways circumvent the vacant *cus site so that the limiting free-energy span shifts from *cus-OOHot → * cus + O2 to *cus-OHot → *cus-Oot. This alteration in the limiting span explains the enhanced electrocatalytic activity of the Walden pathways on the different IrO2(110) surface configurations.
Identifying Walden-type mechanisms as the dominating pathway for the OER over IrO2(110) can be seen as a paradigm change since, hitherto, this category of mechanisms has been largely overlooked for the modeling of proton-coupled electron transfer steps in electrocatalysis.39,40 While we have provided reasoning for the importance of Walden-type pathways in the OER over IrO2(110) based on energetic considerations in the realm of free-energy diagrams (cf. SI, section 9–12), further evidence for the occurrence of Walden steps is given by Bader charge analysis.60 We determine the charge state of the active Ir site (*cus) in both the mononuclear and mononuclear-Walden mechanisms, depicted in Fig. 4a and Fig. 4b, respectively. In the traditional mononuclear mechanism (cf. Figure 4a), the charge state of the Ir atom at the *cus site undergoes multiple fluctuations during the catalytic cycle, ranging from + 1.47e for the vacant active site, *cus, up to + 1.85e for the *cus-Oot adsorbate. On the other hand, changes in the charge state of the active *cus site are much less pronounced for the Walden pathway (cf. Figure 4b). By defining Qmax as the largest charge span in the catalytic cycle, we obtain Qmax = + 0.38e and + 0.17e for the mononuclear and mononuclear-Walden mechanism mechanisms, respectively. Intriguingly, these spans scale with the values of the activity descriptor Gmax(U = 1.53 V) for the mononuclear and mononuclear-Walden mechanisms, which are 0.41 eV and 0.13 eV, respectively (cf. Figure 4).
Based on Fig. 4, we conclude that the stabilized charge state of the Ir atom at the *cus site in the mononuclear-Walden mechanism is linked to its enhanced activity compared to the traditional mechanism. Therefore, we propose that, besides the ubiquitous assessment of adsorption free energies to approximate the electrocatalytic activity, a span model based on the charge states of the active site in the catalytic cycle can be used to gain further insight into proton-coupled electron transfer steps in energy conversion and storage, and this statement may also hold for catalytic processes beyond the OER.
{kind=link}