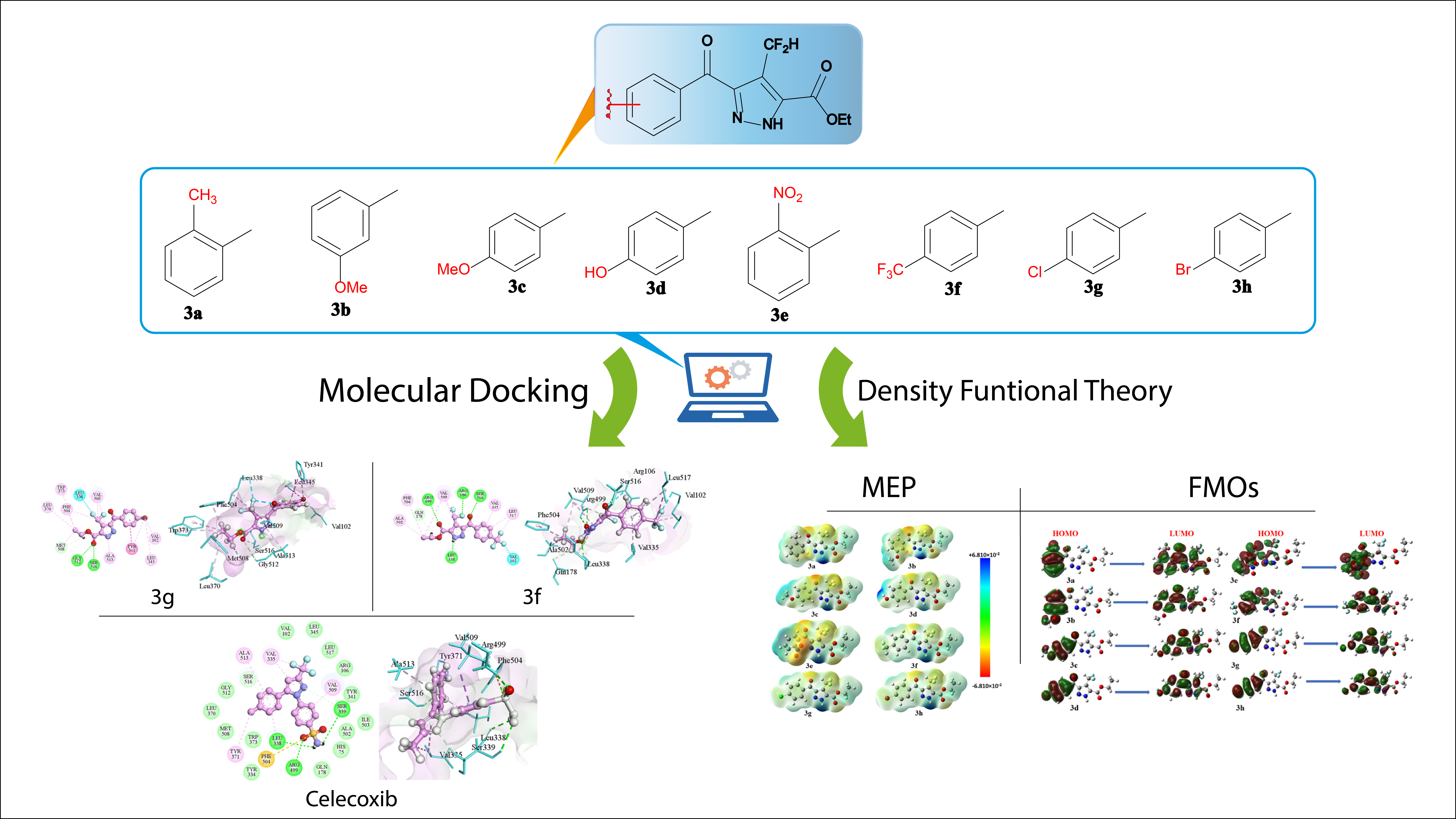
3.1. Molecular docking studies
Molecular docking was performed to investigate the interaction mechanisms and binding strengths of small compounds with the cyclooxygenase enzyme. Utilizing the crystal structure of the musculus target protein with PDB ID 3LN1 as the reference, celecoxib was employed as the ligand in the cocrystal during these investigations. The software MOE was utilized for this docking analysis [28]. A ligand library was compiled using PubChem, and ligands were chosen for the docking studies based on their structural attributes and possible pharmacological significance [29]. The docking analyses revealed the possible binding locations of all ligands to the active site of 3LN1, and the stability of the protein-ligand complexes was also assessed. These results offered a significant understanding of the molecular processes, which controlled the creation of ligand-protein complexes involving protein 3LN1. These findings can also guide additional experimental investigations, such as biochemical testing or structure elucidation, to improve the accuracy of computational predictions and aid in the development of therapeutic drugs or modulators that target protein 3LN1. This study highlights the importance of molecular docking in understanding the binding processes between small drugs and target proteins [30]. Additionally, it establishes the groundwork for the rational development of new compounds with enhanced binding capabilities [31]. Polar hydrogens were added to the macromolecules to prepare them for docking. The MOE Program was used to assign partial atomic charges. The ligand binds directly to the protein at specific positions: Arginine at position 106, Serine at position 516, Tyrosine at position 341, and Leucine at position 338. The binding pattern is influenced by the amino acids i.e., valine (Val) at position 509, phenylalanine (Phe) at position 504, alanine (Ala) at position 502, valine (Val) at position 335, and leucine (Leu) at position 517. The energy score of -7.31191 Cal/mol suggests that the ligand "3a" has a strong binding affinity to the amino acid residues, as indicated by the negative value which signifies stability. The presence of Glutamic acid (Glu) at position 510 and Arginine (Arg) at position 106 is essential for the binding interaction, indicating their importance in maintaining the stability of the ligand-protein complex. Aromatic amino acid residues, such as tyrosine at position 341 and phenylalanine at position 504, could potentially enhance the binding affinity by participating in π-π stacking or other types of interactions. The comprehensive interaction profile of ligand 3b elucidates the precise amino acid residues that facilitate its intricate attachment to the protein, including Alanine (Ala), Arginine (Arg), and Tyrosine (Tyr) at positions 513, 106, and 341 respectively. The energy score of -6.7764 Cal/mol suggests strong binding to the amino acid residues, indicating stability due to its negative value. The presence of Valine at position 509 and Arginine at position 106 is likely to play a crucial role in the process of binding and stability. The presence of aromatic residues, such as Tyrosine at position 341 and Histidine at position 75, may provide complexities in the binding process due to potential π-π stacking or hydrogen bonding interactions. The ligand 3f engages in direct binding interactions with multiple amino acid residues, specifically Arginine, Serine, and Leucine, at positions 499, 106, 516, and 338 respectively. Indirect interactions are observed between amino acids positioned at 504, 502, 509, 335, 517, and 102, along with Glutamine (Gln) at position 178. The interaction score of -7.2866 Cal/mol suggests a robust and advantageous binding affinity, with the negative value reflecting stability. The presence of polar and nonpolar amino acids indicates the participation of different chemical forces, such as hydrogen bonding and hydrophobic interactions. Positions 338, 504, and 178 are identified as crucial for the stabilization of the ligand-protein complex. The ligand 3g directly interacts with amino acids at positions 509, 516, and 106, and indirectly interacts with additional amino acids such as Methionine (Met), Alanine (Ala), Phenylalanine (Phe), Tryptophan (Trp), Leucine (Leu), and Valine (Val). The energy score of -6.9765 Cal/mol suggests a strong and advantageous binding affinity, with the negative number indicating stability. Amino acids at positions 106 and 338 exert a substantial impact on the binding strength, with the presence of aromatic residues suggesting the occurrence of hydrophobic and π-π stacking interactions. The detailed interaction profile provides crucial insights into the chemical mechanisms governing the binding of ligands 3a, 3b, 3f, and 3g to the protein. This information serves as a foundation for further experimental validation and has the potential to guide the development of molecules with enhanced binding characteristics for medication development and molecular design techniques. Figure 2 and Fig. 3 depict the interactions of compounds (3a-3h) with the active pockets of 3LNI amino acid residues in 2D and 3D designs, respectively. Additionally, the interaction of Celecoxib (cognate ligand) with the active pocket of 3LNI amino acid residues is illustrated in both 2D and 3D formats in Fig. 4.
Figure 4
Interaction of Celecoxib (cognate ligand) with an active pocket of 3LNI amino acid residues in 2D and 3D illustration
We evaluated protein target interactions and ligand docking scores as displayed in Table 1. It provided the docking scores (Cal/mol) and contact types, including hydrogen bonds, π-alkyl, π-cation, C-H/π-sigma, and halogen interactions, for ligands and protein targets.
Table 1
The evaluated docking scores (Cal/mol) and contact types (hydrogen bonds, π-alkyl, π-cation, C-H/π-sigma, and halogen) for ligands and protein targets
|
Code
|
Hydrogen
Bonds
|
\(\varvec{\pi }\)-alkyl
|
\(\varvec{\pi }-\varvec{c}\varvec{a}\varvec{t}\varvec{i}\varvec{o}\varvec{n}\)
|
\(\varvec{C}-\varvec{H}/ \varvec{\pi }-\varvec{s}\varvec{i}\varvec{g}\varvec{m}\varvec{a}\)
|
Halogen
|
Docking score
Cal/mol
|
|
3a
|
Arg 106
Ser 516
Tyr 341
Leu 338
|
Val 509
Phe 504
Ala 502
Val 335
Ala 513
Leu 517
|
Arg 106
|
Glu 510
Tyr 341
|
|
-7.31191
|
|
3b
|
Ala 513
Arg 106
Tyr 341
|
His 75
Leu 35
Ala 513
Val 102
Leu 517
|
Arg 106
|
Val 509a
|
|
-6.7764
|
|
3c
|
Arg 106
Arg 106
|
Ala 502
Ala 513
Phe 504
Val 102
Leu 517
Tyr 341
|
Arg 106
|
Ser 339
Gln 178
Val 509
|
|
-6.8477
|
|
3d
|
His 106
Tyr 341
|
Val 102
Val 335
Val 509
Tyr 341
His 75
Ala 502
|
|
|
His 75
|
-6.4321
|
|
3e
|
Arg 499
Arg 106
|
Val 335
Val 509
Tyr 341
His 75
Phe 343
Leu 78
Leu 345
Leu 517
|
Arg 499
Glu 510
Arg 106
|
|
Ala 513
Val 102
|
-7.06335
|
|
3f
|
Arg 499
Arg 106
Ser 516
Leu 338
|
Phe 504
Ala 502
Val 509
Val 335
Leu 517
Val 102
Leu 338
|
|
Gln 178
|
Val 102
|
-7.2866
|
|
3g
|
Val 509
Ser 516
Arg 106
|
Met 508
Ala 513
Phe 504
Trp 373
Leu 370
Val 335
Leu 517
Leu 345
Val 102
|
Arg 106
|
|
Leu 338
|
-6.9765
|
|
3h
|
Gly 512
Ser 516
|
Leu 370
Phe 504
Phe 504
Val 509
Ala 513
Leu 345
Val 102
|
|
Tyr 341
|
Leu 338
|
-6.6180
|
|
Celecoxib
|
Arg 499
Leu 338
Ser 339
|
Ala 513
Val 335
Val 509
Tyr 371
|
Phe 504
|
Ser 516
Ser 339
|
|
-6.9453
|
|
3.2. Frontier molecular orbitals (FMOs)
Frontier molecular orbitals (FMOs) consist of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). The HOMO, encompassing the highest energy level occupied by electrons, signifies an entity with the capacity to donate electrons. In contrast, the LUMO, representing the lowest energy orbital capable of receiving electrons, characterizes a species prepared to accept electrons [32]. These orbitals play a crucial role in dictating how pharmaceuticals engage with other molecules, influencing interactions between these medications and their respective receptors. The frontier molecular orbitals (FMOs) of compounds (3a-3h) provide reliable qualitative insights into the electron transfer susceptibility from HOMO to LUMO, as depicted in Fig. 5. Furthermore, the HOMO and LUMO play a crucial role as quantum chemical parameters in assessing the reactivity of molecules. They are utilized in the computation of several significant characteristics, including chemical reactivity parameters. The HOMO and LUMO energies of the compounds under investigation were determined using Gaussian at PBE0-D3BJ approach with a def2-TZVP basis set. The calculated electronic properties of the compounds (3a-3h), such as EHOMO, ELUMO, and energy gap (∆E) between ELUMO and EHOMO values are displayed in Table 2.
Table 2
Calculated electronic properties of the compounds (3a-3h), such as EHOMO, ELUMO, and energy gap (∆E)
|
Compounds
|
EHOMO (eV)
|
ELUMO (eV)
|
∆E = ELUMO - EHOMO (eV)
|
|
3a
|
-7.12
|
-1.83
|
5.28
|
|
3b
|
-6.52
|
-1.92
|
4.59
|
|
3c
|
-6.63
|
-1.79
|
4.83
|
|
3d
|
-6.74
|
-1.82
|
4.92
|
|
3e
|
-7.76
|
-2.44
|
5.32
|
|
3f
|
-7.74
|
-2.30
|
5.44
|
|
3g
|
-7.28
|
-2.13
|
5.14
|
|
3h
|
-7.21
|
-2.17
|
5.04
|
HOMO and LUMO are influential in determining various molecular properties, including optical and electrical properties, chemical reactivity and stability, biological activity arising from intermolecular charge transfer, chemical hardness, and chemical softness. HOMO energy refers to the molecule's capacity to donate electrons, while LUMO energy refers to the molecule's capacity to accept electrons. The energy difference between HOMO and LUMO provides insights into the molecule's stability [33]. A substantial energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) signifies diminished reactivity, indicating the stability of the molecule [34].
3.3. Molecular electrostatic potential evaluation
The MEP graph of a molecule depicts how the positive point charge interacts with the charges scattered among its atoms. Although the areas highlighted in red on both maps represent the region vulnerable to chemical reactions, where the electron density is larger than the nucleus throughout the entire molecule, it signifies the negative region of the electrostatic potential. The partial positive charge and unstable reaction zones are indicated by the blue areas on the potential surface. Yellow color indicates lesser electron density, whereas green color denotes nearly neutral areas [35]. The MEP map of 3a-3h compounds showed that the perimeter of the carbon and hydrogen atoms seemed more positively charged, but the red zones were primarily concentrated on the oxygen atoms, which are more negatively charged.
Molecular Electrostatic Potential (MEP) is employed to predict the locations of nucleophilic and electrophilic attacking sites within a molecule, providing insights into their relative reactivity. The MEP analysis was conducted on the optimized structures of all compounds. The molecular electrostatic potential (MEP) can be a useful method for verifying data, indicating that these compounds interact as inhibitors. MEP uses color categorization to define a molecule’s size, shape, positive, negative, and neutral areas. Red, orange, yellow, green, and blue are the colors in ascending order of potential [36]. By following the ring arrangement, it is quite simple to locate places that are suitable for nucleophile and electrophile attack. In contrast to the red color representing regions of low electrostatic potential, indicating an abundance of electrons, the blue color denotes regions of extreme electrostatic potential. This designation signifies the non-existence of electrons in this region, making it a preferred site for nucleophilic attack [37]. The oxygen atoms of the nitro group (red coded area) in each system are assigned regions of minimal potential following the MEP analysis, as illustrated in Fig. 6.
3.4. Conceptual DFT reactivity parameters
The reactivity parameters of the molecules are calculated from the total energies and the Koopmans’ theorem. The ionization potential i.e., \({IP}_{TE}\) and \({IP}_{KE}\) are determined from the energy difference between the energy of the compound derived from electron transfer (radical caution) and the respective neutral compound and negative of EHOMO respectively, as provided by Eqs. (1) and (2)
|
\({IP}_{TE}={E}_{cation}-{E}_{n}\)
|
(1)
|
|
\({IP}_{KE}=-{E}_{HOMO}\)
|
(2)
|
The electron affinity i.e., \({EA}_{TE}\) and \({EA}_{KE}\) are computed from the energy difference between the neutral molecule and the anion molecule and negative of LUMO energy, as given in Eqs. (3) and (4) respectively [38].
$${EA}_{TE}={E}_{n}-{E}_{anion}$$
3
$${EA}_{KE}=-{E}_{LUMO}$$
4
The other important quantities such as chemical potential (µ), hardness (ƞ), and electrophilicity index (ω) were calculated from the following Eq. (5)(6)(7) [39].
|
\({\eta } = (\text{I}–\text{A})/2\)
|
(5)
|
|
\({\mu } = - (\text{I}+\text{A})/2\)
|
(6)
|
|
\({\omega } = {\mu }2/2{\eta }\)
|
(7)
|
The Table 3 presents the values of reactivity parameters i.e., IP (ionization potential), EA (electron affinity), ƞ (chemical hardness), µ (chemical potential), and ω (electrophilicity) in eV of the selected compounds (3a-3h).
Table 3
The table presents the calculated chemical parameters of the selected compounds (3a-3h)
|
Compounds
|
IP (eV)
|
EA (eV)
|
ƞ (eV)
|
µ (eV)
|
ω (eV)
|
|
3a
|
7.12
|
1.83
|
2.64
|
-4.47
|
3.79
|
|
3b
|
6.52
|
1.92
|
2.29
|
-4.22
|
3.88
|
|
3c
|
6.63
|
1.79
|
2.41
|
-4.21
|
3.67
|
|
3d
|
6.74
|
1.82
|
2.46
|
-4.28
|
3.72
|
|
3e
|
7.76
|
2.44
|
2.66
|
-5.10
|
4.89
|
|
3f
|
7.74
|
2.30
|
2.72
|
-5.02
|
4.63
|
|
3g
|
7.28
|
2.13
|
2.57
|
-4.70
|
4.30
|
|
3h
|
7.21
|
2.17
|
2.52
|
-4.69
|
4.36
|
A detailed analysis was conducted to examine the electronic characteristics of a set of 4-difluoromethyl pyrazole derivatives (3a-3h), which revealed clear distinctions in energy-related metrics. The compounds displayed a range of ionization potentials (IP) from 6.52 eV to 7.76 eV, indicating their tendency to donate electrons [40]. Additionally, the electron affinities (EA) exhibited a range from 1.79 eV to 2.44 eV, providing valuable insights into the compounds' ability to accept electrons. The variations in the HOMO-LUMO energy gaps (ƞ) within the range of 2.29 eV to 2.72 eV indicated electronic stability and reactivity among the compounds. Moreover, the dipole moments (µ) ranged from − 4.47 eV to -5.10 eV, implying the polarity of the compounds and their potential for interactions with other molecules [41]. Finally, the electronegativities (ω) ranged from 3.67 eV to 4.89 eV, indicating the compounds' varying capacities to attract electrons [42]. The detailed electronic characterizations significantly contribute to our understanding of the distinct attributes of each derivative. This enhanced knowledge not only facilitates further exploration of their potential biological activities but also plays a crucial role in strategically developing new compounds tailored for specific medicinal purposes.
{kind=link}