Recombinant proteins and fibril formation
Human WT α-synuclein protein and AS69 were produced in bacteria and purified as previously described [10]. In the final purification step, WT aSyn was eluted from a Superdex 75 column (GE Healthcare) equilibrated in PBS buffer. PFF were generated using a standard protocol [8]. In brief, 500 µl of a 5 mg/ml solution of aSyn in PBS were incubated in 1.5 ml microcentrifuge tubes on a Thermomixer (Eppendorf) sealed with parafilm for 7 days at 37 °C under constant agitation (1000 rpm). Fibril formation was confirmed by Thioflavin T fluorescence and atomic force microscopy.
Animals and surgery
C57BL6/J-Thy1-A30P-α-synuclein mice [9] have been used in previous studies [11–13]. They were bred as homozygous, housed and handled in a pathogen-free animal facility at 20–24 °C with a 12 h light/dark cycle, food and water ad libitum, in accordance with guidelines of the Federation for European Laboratory Animal Science Associations (FELASA). Breeding and surgery were approved by the local authorities (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, licence numbers 84.02.04.2015.A027 and 84-02.04.2014.A321).
Mice were identified by three-digit numbers and assigned to one of three experimental groups using random numbers by a technician not involved in the design of the study. The injected solution was prepared freshly on each experimental day from frozen aliquots of the following three stock solutions: sterile phosphate buffer solution (PBS), PFF in PBS at a concentration corresponding to 5 mg/ml aSyn protein, 404 µM AS69 in PBS. After thawing, PFF were diluted in PBS in order to obtain the same PFF concentration both with and without AS69. The final solutions contained (1) PBS only, (2) 1.4 mg/ml α-synuclein equivalent PFF, (3) 1.4 mg/ml α-synuclein equivalent PFF + 98 µM AS69. (98 µM AS69 is roughly equimolar to 1.4 mg/ml α-synuclein.) After dilution, solutions were sonicated at room temperature with 10% power output and 60 pulses of one second each (300 VT; Biologics, Inc., Manassas, VA).
Stereotaxic injection into the right striatum (AP: 1; ML: 1.5 relative to Bregma; DV: 1.55 from dura) and tissue preparations were performed essentially as described earlier [12]. Briefly, 47–57 week old mice (mixed gender) received 2.5 µl of one of the three solutions (PBS, PFF, PFF + AS69) with a flow rate of 0.2 µl/min. Mice were sacrificed 90 days after the injection. Brains were extracted, fixed at 4 °C in 4% paraformaldehyde in PBS for 24 hours and cryoprotected in 30% sucrose in PBS. Free-floating, 30 µm serial coronal sections were collected with a Cryostat (Leica Microsystems, Germany). Brain slices were stored at − 20 °C (30% glycerol, 30% ethylene glycol in 0.1 M PB) until use.
Primary neuronal culture
Primary neuronal cultures were prepared from P1-3 C57BL6/J mouse pups of mixed sex. Briefly, after dissection and trypsinisation, dissociated neurons were plated onto poly-L-ornithine (Sigma) coated glass coverslip (100 000 cells per well in 24-well dishes), and maintained in Neurobasal A medium containing B27 (2%, Invitrogen), glutamax (0.5 mM, Invitrogen) and antibiotics [14]. One third of the medium was changed on every third day, and from the second change, no antibiotics was added. On DIV 12, neurons were incubated with BSA (1 µg/ml, protein control), 50 nM AS69, PFF corresponding to 50 nM aSyn monomer or the same concentration of PFF + 50 nM AS69. Neurons were analysed on day 13 (24 h after adding PFF) or day 15 (72 h after adding PFF). Experiments were repeated 3–4 times (n = 3–4).
Immunostaining
To visualize the α-synuclein pathology, every 6th section of the striatum was first incubated in 0.3% H2O2 for 30 minutes to block endogenous peroxidase activity, then in 3% normal goat serum for 60 min. Sections were then incubated with a primary anti-phospho-synuclein antibody (rabbit, 1:500, ab51253, Abcam, overnight, 4 °C) in 3% normal goat serum, followed by incubation with biotinylated secondary antibody (goat anti-rabbit IgG, 1:200, Vector Laboratories BA-1000), and then with Avidin-Biotin Complex (1:100, Vectastain ABC-Kit Standard PK-6100, Vector Laboratories) for 30 minutes at 21 °C each. Antibody labelling was visualized by exposure to 4 mg/ml 3,3′ diaminobenzidine (DAB) (Sigma Aldrich), and 1% (v/v) H2O2 in Tris buffer. Sections were mounted on glass slides, counterstained with Hemalaun, dehydrated to Xylene and coverslipped with Entellan (Merck).
To determine density of dopaminergic axon terminals in the striatum, three sections spanning Bregma levels + 0.26 to + 0.98 mm [15] per animal were stained for TH [16]. After blocking (3% normal goat serum, 60 min), sections were incubated in the presence of the primary TH antibody (rabbit; 1:1000, AB152, Merck Millipore, overnight, 4 °C) followed by secondary antibody (Alexa 488 conjugated goat anti-rabbit; 1:1000, Invitrogen, Carlsbad, CA, USA, 60 min). Sections were mounted with Fluoromount-G (Southern Biotech, Birmingham, AL, USA).
To determine neuroinflammation, every sixth striatal sections were incubated in the presence of the astroglia marker GFAP (chicken, 1:2000, ab4674, Abcam) and for the microglia marker Iba1 (rabbit, 1:1000, 019-19741, Wako, overnight, 4 °C). After incubation with fluorescently labelled secondary antibodies (Alexa 488 conjugated goat anti-chicken and Alexa 555 conjugated donkey anti-rabbit, 1:1000, 120 min), sections were counterstained with Hoechst (Thermo Fischer) and mounted with Fluoromount-G. To determine the density of dopaminergic (TH positive) neurons in the substantia nigra, every third section was stained for TH as described for anti-phospho-synuclein (i.e. with DAB), but using the primary TH antibody (1:1000, overnight, 4 °C).
To determine aSyn uptake and pathology in primary neuronal cultures, fixed cells were permeabilized (0.2% Triton X-100), unspecific sites were blocked (2% BSA), and incubated in the presence of the following primary antibodies (4 °C, overnight): anti phospho-aSyn (mouse, 1:1000, 015-25191, Wako), anti human-aSyn (rat, 15G7, 1:500, ALX-804-258-L001, Enzo Life Sciences), anti-affibody (goat, 1:500, Ab50345, Abcam), anti-MAP2 (rabbit, 1:1000, AB5622, Merck). After incubation with fluorescently labelled secondary antibodies (Alexa 405 conjugated donkey anti-rabbit, anti-Alexa 488 conjugated donkey anti-rat, Alexa 555 conjugated donkey anti-goat and Alexa 647 conjugated donkey anti-mouse), coverslips were mounted on glass objective slides (Fluoromount G).
Analysis of aSyn pathology, striatal fiber density and gliosis
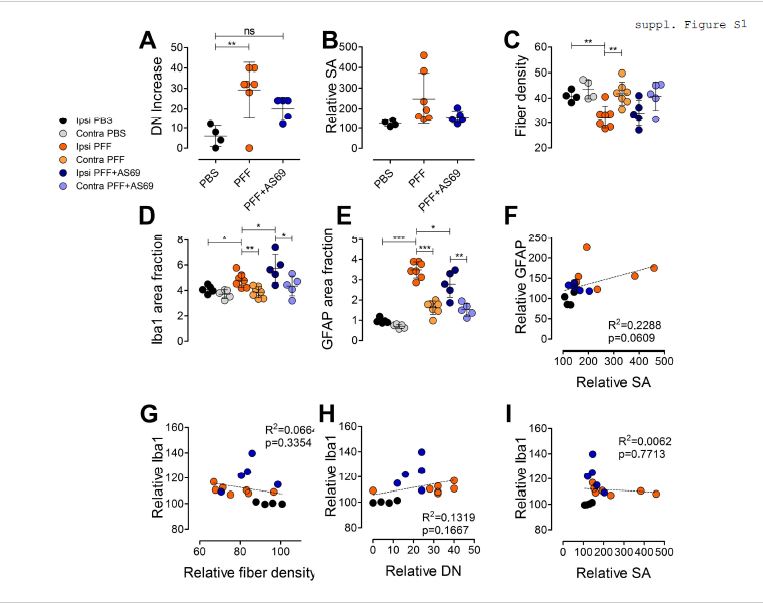
To quantify the extent of aSyn pathology in the striatum, we used the StereoInvestigator software (MicroBrightfield Bioscience, Williston, VT) and the optical fractionator method. Every 6th slice of the striatum was stained. In each slice, a grid of 200 µm × 200 µm was placed randomly over the striatum outlined using the 2.5x objective. At each intersection of the grid, a 100 µm × 100 µm counting frame was inspected manually for phospho-aSyn positive somatic inclusions and dystrophic neurites using a 63x oil objective and an Axio Imager 2 microscope (Carl Zeiss Vision, Göttingen, Germany). The investigator carrying out this analysis was not involved in the surgery and unaware of the injection obtained by each animal (identified by three-digit numbers). Counts reflect the number of items estimated by the stereology procedure for each animal.
For quantification of the density of striatal TH-positive fibers, fluorescence 8-bit images were acquired as z-stack (five slices, 1 µm step) using a 60x oil objective (NA 1.35) and an IX81S1F microscope (Olympus, Hamburg, Germany). A maximum intensity projection over z was performed and TH-positive fibres delineated using the auto-threshold function of ImageJ (1.47v; NIH, Bethesda, MD, USA). Fiber density was expressed as percent area. Three section per animal, and five images per section were analysed resulted in a hierarchically nested design. A generalized linear mixed model was applied [13].
For quantification of gliosis, fluorescent images were acquired using a 20x objective (NA 0.8) with an Axio Imager 2 microscope (Zeiss). After adjusting the threshold on the single images (separate for GFAP and for Iba1; ImageJ), area fraction was determined from 2 sections per animal and from 10 images per section (see above).
The number of dopaminergic neurons in the substantia nigra was quantified as previously [16]: TH-positive neurons in the SNc of the right hemisphere were stereologically counted using the optical fractionator method (StereoInvestigator) in every third section (counting frame: 100 × 100 µm, grid size; 200 × 200 µm; oil immersion 63x objective, NA 1.25).
To measure aSyn uptake and pathology, images of primary cultures were acquired with Zeiss Axio Imager 2 using a 100x oil immersion objective (NA 1.4), with a monochrome camera (Axiocam 705 mono, Zeiss). For each experiment and experimental group, a minimum of 10 neurons were selected randomly. Exposure time was kept constant for each staining across all experimental groups. To outline neurons, a mask was created based on the MAP2 channel. In this mask, the area fraction of (a) phospho-aSyn and (b) human aSyn staining was determined, using ImageJ 1.52 h. Images are pseudo coloured for better visualization.
Statistical analysis and data visualisation
For cell cultures, “n” was set to the number of individual experiments, and for the animal experiments, “n” was set to the number of mice. For each experimental condition and experiment, or for each animal, values were summarized by determining the mean. Data are presented as markers for each condition within a single experiment, and for each animal and as mean ± standard deviation. Comparisons were performed one-way or two-way ANOVA using GraphPad Prism 5 (Version 5.01). p < 0.05 was considered significant. p values are depicted as * p < 0.05; ** p < 0.01.
{kind=link}